Home
Last updated: 2023-02-13
Checks: 1 1
Knit directory: XMAP-tutorial/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version c276160. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rproj.user/
Unstaged changes:
Modified: analysis/_site.yml
Modified: analysis/index.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/index.Rmd) and HTML
(docs/index.html) files. If you’ve configured a remote Git
repository (see ?wflow_git_remote), click on the hyperlinks
in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | c276160 | mxcai | 2023-02-13 | Start workflowr project. |
Overview
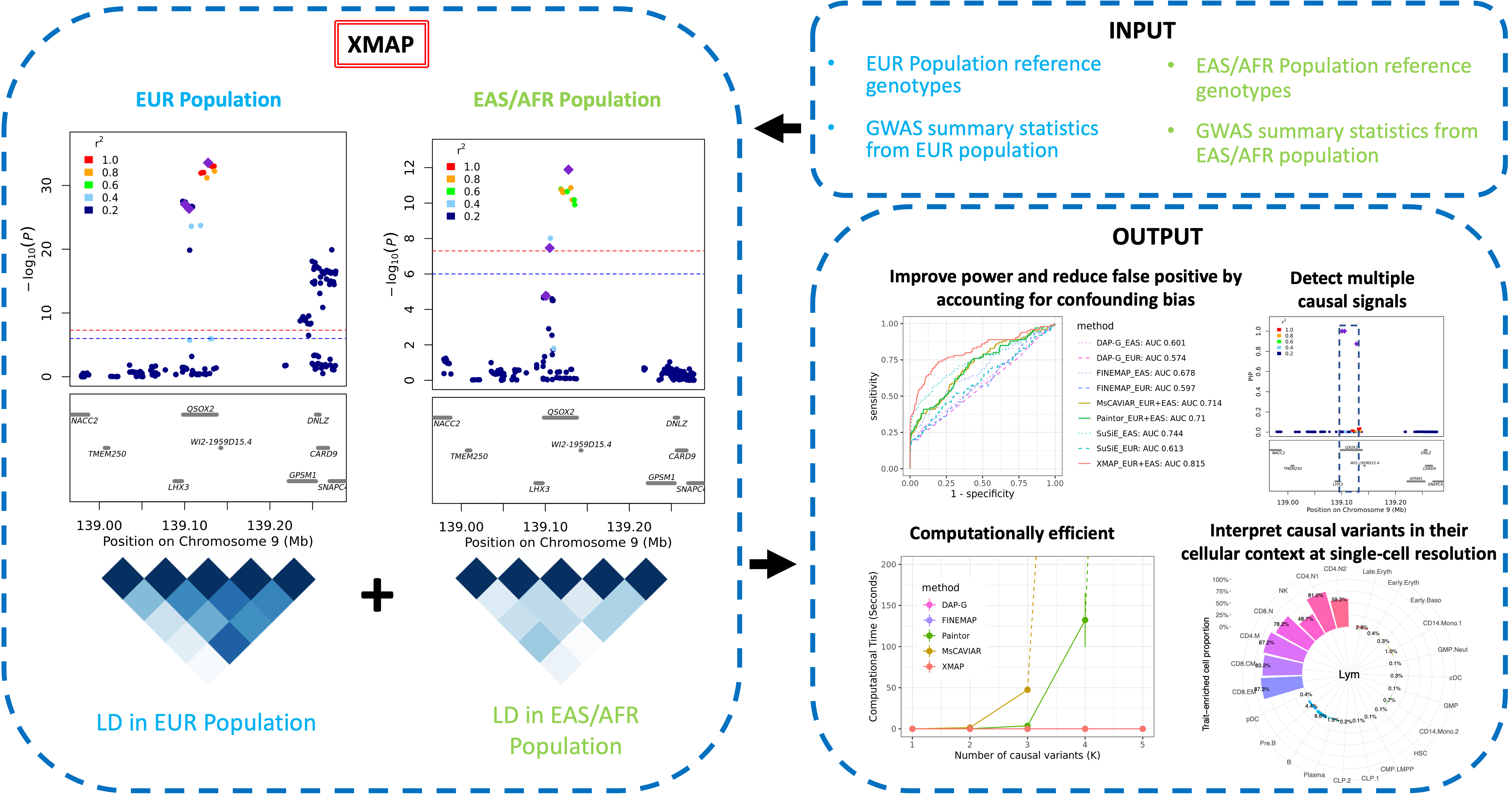
XMAP is a computationally efficient and statistically accurate method for fine-mapping causal variants using GWAS summary statistics. Briefly, it can leverage different LD structures of genetically diverged populations to better distinguish causal variants from a set of associated variants. By jointly modeling SNPs with putative causal effects and polygenic effects, XMAP allows a linear-time computational cost to identify multiple causal variants, even in the presence of an over-specified number of causal variants. It further corrects confounding bias hidden in the GWAS summary statistics to reduce false positive findings and improve replication rates.
The fine-mapping results given by XMAP can be further used for downstream analysis to illuminate the causal mechanisms at different cascades of biological processes, including tissues, cell populations, and individual cells. In particular, XMAP results can be effectively integrated with single-cell datasets to identify disease/trait-relevant cells.