Trypanosoma phyloseq
Last updated: 2021-07-02
Checks: 7 0
Knit directory: wildlife-haemoprotozoa/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210212) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version b1f7e30. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: data/.DS_Store
Ignored: output/.DS_Store
Untracked files:
Untracked: ENA_submission/
Untracked: NCBI_submission/
Untracked: Rplot02.pdf
Untracked: data/NGS_18S_kinet/
Untracked: data/Rdata/tryp_amp.RData
Untracked: data/Sanger-piroplasm/
Untracked: data/haemoprotozoa.csv
Untracked: data/haemoprotozoa.xlsx
Untracked: data/tryp-phyloseq/count_data_QC.xlsx
Untracked: data/tryp-phyloseq/count_data_raw.csv
Untracked: data/tryp-phyloseq/sampledata.xlsx
Untracked: melt.csv
Untracked: melt.xlsx
Untracked: microscopy/
Untracked: output/plots/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/tryp-02-phyloseq.Rmd) and HTML (docs/tryp-02-phyloseq.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | b1f7e30 | siobhon-egan | 2021-07-02 | include main figs and tables |
| html | a36fbcb | siobhon-egan | 2021-07-02 | Build site. |
| Rmd | 8e3850e | siobhon-egan | 2021-07-02 | Major update |
Visualization and analysis of trypanosome NGS data data.
Note this was run using R version 4.0.3 and RStudo version 1.4. See R info for full details of R session.
0. Load libraries
Install libraries if required
Only need to run this code once.
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
# phyloseq
source('http://bioconductor.org/biocLite.R')
biocLite('phyloseq')
#tidyverse
install. packages("tidyverse")
#ampvis2
install.packages("remotes")
remotes::install_github("MadsAlbertsen/ampvis2")
#ampvis2extras
install.packages("BiocManager")
BiocManager::install("kasperskytte/ampvis2extras")
#ggpubr
install.packages("ggpubr")
#agricolae
install.packages("agricolae")
install.packages("remotes")
remotes::install_github("DanielSprockett/reltools")
devtools::install_github('jsilve24/philr')
#decontam
BiocManager::install("decontam")
library(decontam)
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("Biostrings")Load libraries
pkgs <- c("qiime2R", "phyloseq", "tidyverse", "ampvis2",
"ampvis2extras", "ggpubr", "agricolae", "plotly",
"viridis", "cowplot", "MicrobeR", "microbiome",
"reshape", "decontam", "data.table", "ape",
"DESeq2", "vegan", "microbiomeutilities", "knitr",
"tibble", "dplyr","patchwork", "Biostrings")
lapply(pkgs, require, character.only = TRUE)
theme_set(theme_bw())1. Import data
Generate phyloseq object from spreadsheets.
Import ASV/OTU count data
count_data <- read_csv("data/tryp-phyloseq/count_data_cleaned.csv")
# use first column as label for rows
count_data_lab = column_to_rownames(count_data, var = "#Zotu ID")
# Make matrix
otumat <- as.matrix(count_data_lab)Import taxonomy data
taxonomy <- read_csv("data/tryp-phyloseq/taxonomy.csv",
col_types = cols(Accession_description = col_skip(),
`Accession no.` = col_skip(), evalue = col_skip(),
`per. Ident` = col_skip(), taxid = col_skip()))
# use first column as label for rows
taxonomy_lab = column_to_rownames(taxonomy, var = "#Zotu ID")
taxmat <- as.matrix(taxonomy_lab)Check the class of the otumat and taxmat objects, they MUST be in matrix format. Then we can great a phyloseq object called physeq from the otu and taxonomy tables and check the sample names.
class(otumat)
class(taxmat)
OTU = otu_table(otumat, taxa_are_rows = TRUE)
TAX = tax_table(taxmat)
physeq = phyloseq(OTU, TAX)
physeq
sample_names(physeq)Add metadata and sequence data
Add sequences to phyloseq object
# read sequence file
rep.seqs <- Biostrings::readDNAStringSet("data/tryp-phyloseq/unoise_zotus.fasta", format = "fasta")Add metadata, importing gDNAID as factor to be able to merge later on
metadata <- read_csv("data/tryp-phyloseq/sampledata.csv")
metadata_lab = column_to_rownames(metadata, var = "SampleID")
sampledata = sample_data(data.frame(metadata_lab))
sampledataCreate final phyloseq object
Now you can merge your data to create a final phyloseq object
ps_raw_tryp = merge_phyloseq(physeq, sampledata, rep.seqs)Preliminary subset
Remove samples with NA values or not part of final data set,
ps_raw_tryp <- subset_samples(ps_raw_tryp, !SampleType=="SampleEcol")
ps_samp_tryp <- subset_samples(ps_raw_tryp, SampleType=="Sample")Save/load .RData
Save R data for phyloseq object - saving “raw data” which inc controls (ps_raw_tryp) and “sample only data” (ps_samp_tryp)
save(ps_raw_tryp, file = "data/Rdata/ps_raw_tryp.RData")
save(ps_samp_tryp, file = "data/Rdata/ps_samp_tryp.RData")To load raw and sample data quickly from .RData format.
load("data/Rdata/ps_raw_tryp.RData")
load("data/Rdata/ps_samp_tryp.RData")An easy way to view the tables is using Nice Tables
Nice.Table(ps_samp_tryp@sam_data)Nice.Table(ps_samp_tryp@tax_table)2. QC plots
R package decontam to assess contaminating OTUs, tutorial.
The CRAN version only works on R version <4. To install for R versions >4 install from bioconductor using the following
Make plot of library size of Samples vs Controls
df <- as.data.frame(sample_data(ps_raw_tryp)) # Put sample_data into a ggplot-friendly data.frame
df$LibrarySize <- sample_sums(ps_raw_tryp)
df <- df[order(df$LibrarySize),]
df$Index <- seq(nrow(df))
libQC <- ggplot(data=df, aes(x=Index, y=LibrarySize, color=SampleType)) + geom_point() + theme_bw() + scale_colour_brewer(palette = "Set1")
libQC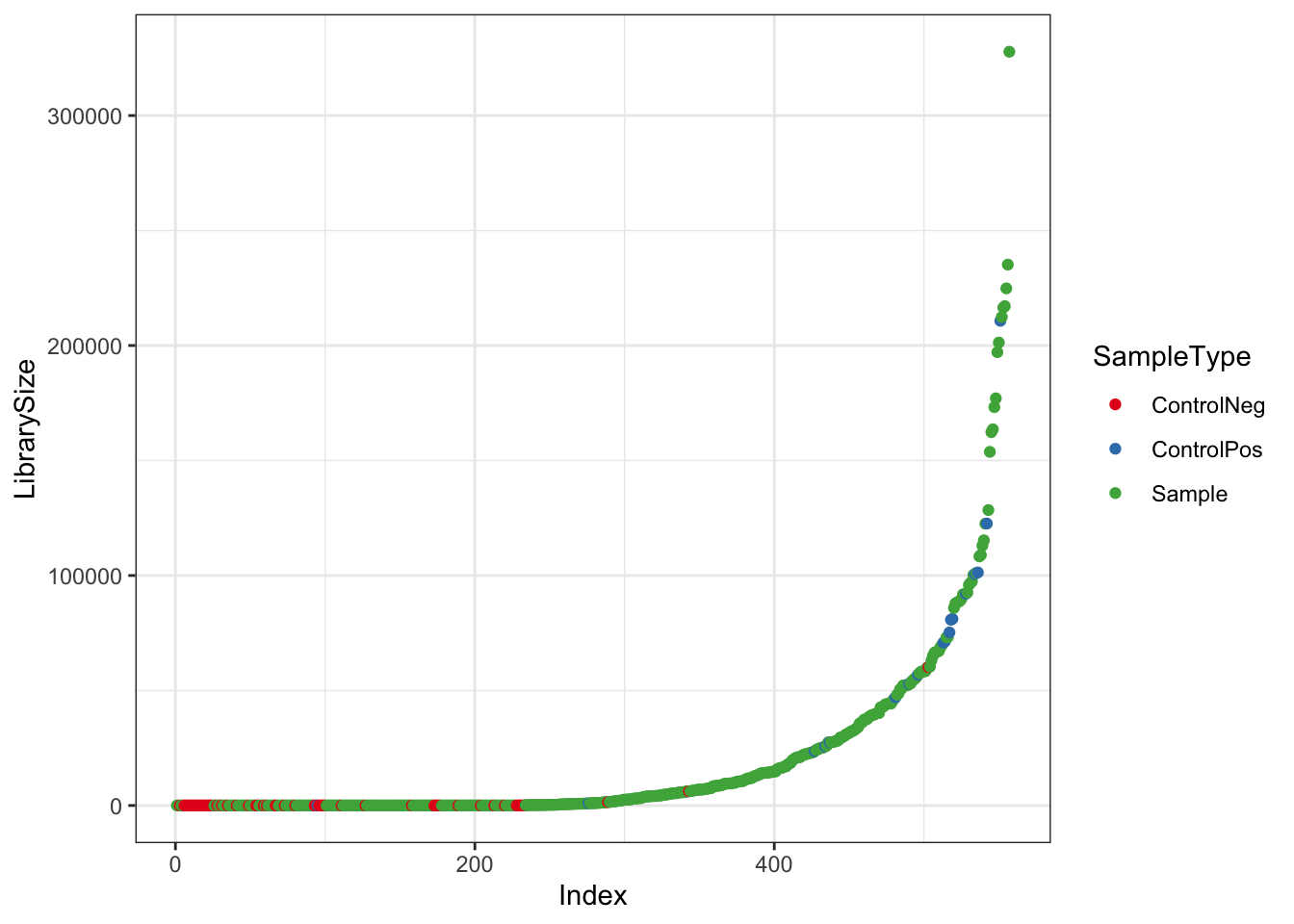
#ggsave("libQC.pdf", plot = libQC, path = "output/plots/trypNGS", width = 15, height = 10, units = "cm")Make html plot with plotly
libQCplotly <- ggplotly(libQC)
libQCplotly#htmlwidgets::saveWidget(libQCplotly, "output/plots/libQCplotly.html")Make distribution plot of reads using microbiomeutilities
distrib <- plot_read_distribution(ps_raw_tryp, groups = "SampleCategory",
plot.type = "density") + xlab("Reads per sample") + ylab("Density")[1] "Done plotting"distrib <- distrib + geom_density(alpha = 0.5, fill = "grey") + theme(axis.text.x = element_text(angle = 45, size=10, vjust = 1))
distrib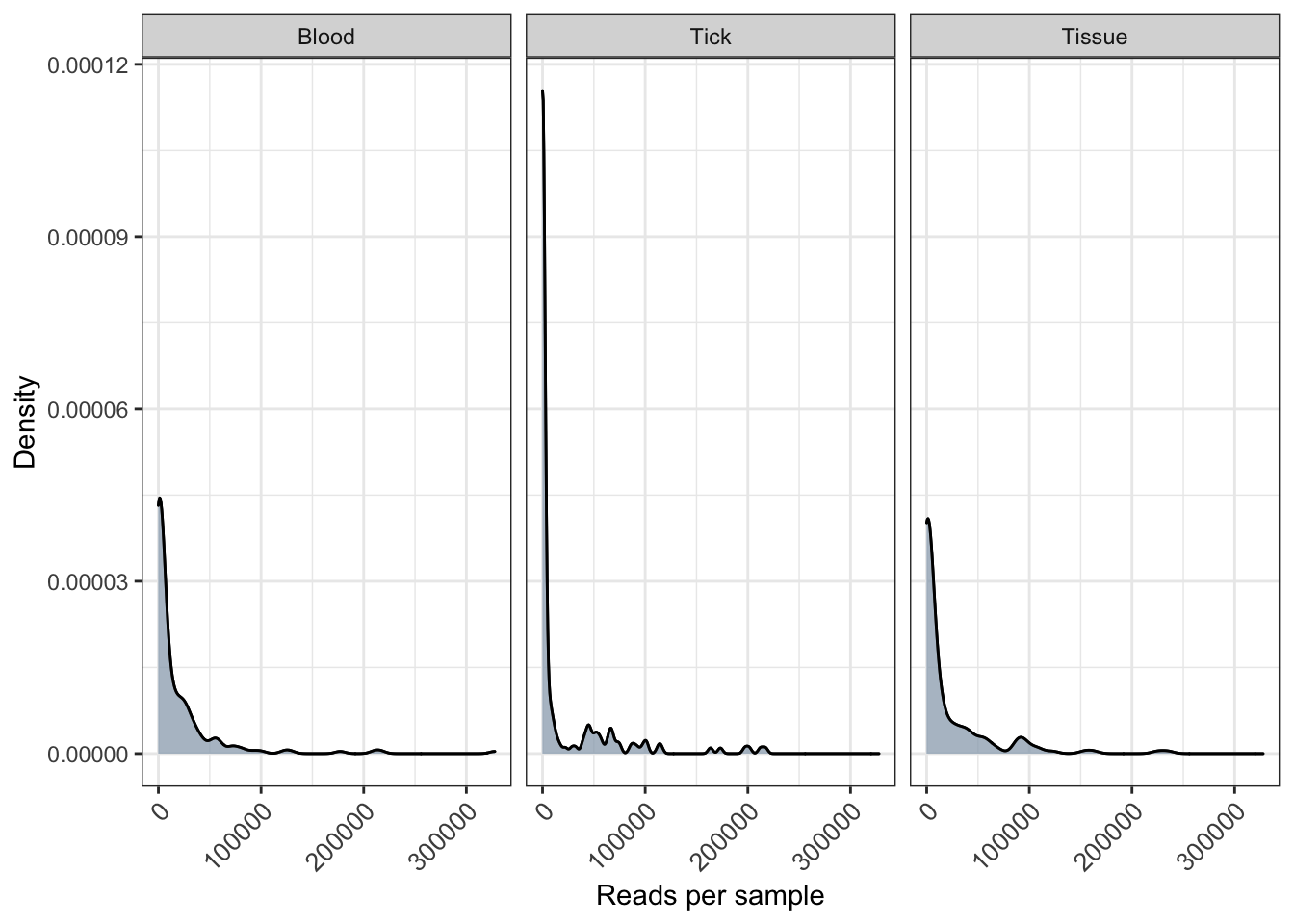
#ggsave("distrib.pdf", plot = distrib, path = "output/plots/trypNGS", width = 15, height = 10, units = "cm")Merge the two above plots into one figure.
QC <- ggarrange(libQC, distrib,
labels = c("A", "B"),
ncol = 1, nrow = 2)
#ggsave("QC.pdf", plot = QC, path = "output/plots/trypNGS", width = 20, height = 20, units = "cm")3. Subset and Filter
3.1. For phyloseq object
Subset phyloseq object based on sample types
# samples and positive controls
ps_tryp_sampcon = subset_samples(ps_raw_tryp, SampleType=="Sample" | SampleType=="ControlPos")
# Blood samples only
ps_tryp_bl = subset_samples(ps_samp_tryp, SampleCategory=="Blood")
# Tissue samples only
ps_tryp_tis = subset_samples(ps_samp_tryp, SampleCategory=="Tissue")
# Tick samples only
ps_tryp_tick = subset_samples(ps_samp_tryp, SampleCategory=="Tick")Subset phyloseq object based on host species
# Black rat
ps_BR = subset_samples(ps_samp_tryp, species=="Black rat")
# Brush tail possum
ps_BTP = subset_samples(ps_samp_tryp, species=="Brush tail possum")
# Chuditch
ps_chud = subset_samples(ps_samp_tryp, species=="Chuditch")
# Long-nosed bandicoot
ps_LNB = subset_samples(ps_samp_tryp, species=="Long-nosed bandicoot")3.2. For ampvis2 object
Make ampvis2 object for analysis
#require the devtools package to source gist
if(!require("devtools"))
install.packages("devtools")
#source the phyloseq_to_ampvis2() function from the gist
devtools::source_gist("8d0ca4206a66be7ff6d76fc4ab8e66c6")
#convert
tryp_amp <- phyloseq_to_ampvis2(ps_samp_tryp)
#save ampvis2 RData obj
save(tryp_amp, file = "data/Rdata/tryp_amp.RData")Load in saved ampvis2 obj
load("data/Rdata/tryp_amp.RData")Subset ampvis2 object based on sample category
#remove controls
amp_samp <- amp_subset_samples(tryp_amp,
!SampleType %in% c("ControlPos"),
RemoveAbsents = TRUE)0 samples have been filtered.#blood samples
amp_bl <- amp_subset_samples(amp_samp,
SampleCategory %in% c("Blood"),
RemoveAbsents = TRUE)316 samples and 410 OTUs have been filtered
Before: 477 samples and 587 OTUs
After: 161 samples and 177 OTUs#tissue samples
am_tis <- amp_subset_samples(amp_samp,
SampleCategory %in% c("Tissue"),
RemoveAbsents = TRUE)321 samples and 214 OTUs have been filtered
Before: 477 samples and 587 OTUs
After: 156 samples and 373 OTUs#tick samples
amp_tick <- amp_subset_samples(amp_samp,
SampleCategory %in% c("Tick"),
RemoveAbsents = TRUE)317 samples and 512 OTUs have been filtered
Before: 477 samples and 587 OTUs
After: 160 samples and 75 OTUs4. Heatmap
4.1. ampvis2
Make heat map using ampvis2.
First filter taxa
# order Trypanosomatida
tax_vector1 <- c(
"Trypanosomatida"
)
amp_samp_otry <- amp_subset_taxa(amp_samp,
tax_vector = tax_vector1)Warning: One or more samples have 0 total reads542 OTUs have been filtered
Before: 587 OTUs
After: 45 OTUs# Trypanosoma species of interest
tax_vector2 <- c(
"Trypanosoma gilletti",
"Trypanosoma sp. (cyclops-like)",
"Trypanosoma vegrandis",
"Trypanosoma noyesi",
"Trypanosoma sp. (lewisi-like)",
"Lotmaria passim"
)
amp_samp_targettryp <- amp_subset_taxa(amp_samp,
tax_vector = tax_vector2)Warning: One or more samples have 0 total reads544 OTUs have been filtered
Before: 587 OTUs
After: 43 OTUsRelative abundance
Heatmap of subsetted data using relative abundance
# Relative abundance order level trypanosome subset obj
heatmap_rel1 <- amp_heatmap(amp_samp_otry,
facet_by = "SampleCategory",
group_by = "species",
tax_aggregate = "Species",
tax_show = 10,
normalise = TRUE,
plot_values = FALSE,
plot_values_size = 3,
round = 0, color_vector = c("white", "#e5ba52", "#ab7ca3", "#9d02d7", "#0030bf"), plot_colorscale = "log10") +
theme(axis.text.x = element_text(angle = 45, size=10, vjust = 1),
axis.text.y = element_text(size=10),
legend.position="right")Warning: There are only 7 taxa, showing allheatmap_rel1Warning: Transformation introduced infinite values in discrete y-axis
# Relative abundance species level trypanosome subset obj
heatmap_rel2 <- amp_heatmap(amp_samp_targettryp,
facet_by = "SampleCategory",
group_by = "species",
tax_aggregate = "Species",
tax_show = 10,
normalise = TRUE,
plot_values = FALSE,
plot_values_size = 3,
round = 0, color_vector = c("white", "#e5ba52", "#ab7ca3", "#9d02d7", "#0030bf"), plot_colorscale = "log10") +
theme(axis.text.x = element_text(angle = 45, size=10, vjust = 1),
axis.text.y = element_text(size=10),
legend.position="right")Warning: There are only 6 taxa, showing allheatmap_rel2Warning: Transformation introduced infinite values in discrete y-axis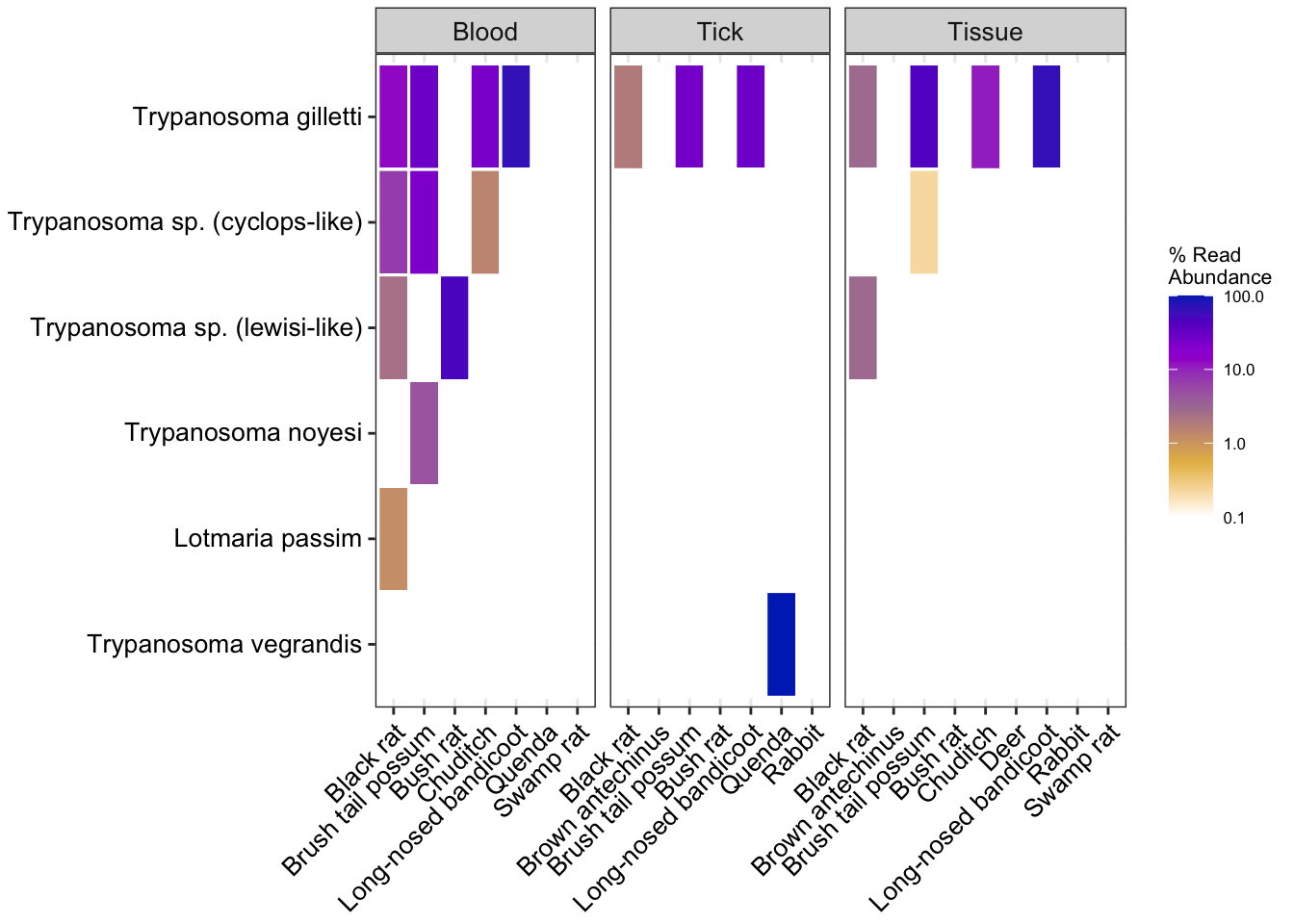
Save PDF of plots
ggsave("heatmap_rel1.pdf", plot = heatmap_rel1, path = "output/plots/trypNGS", width = 25, height = 15, units = "cm")
ggsave("heatmap_rel2.pdf", plot = heatmap_rel2, path = "output/plots/trypNGS", width = 25, height = 15, units = "cm")Count of sequences
heatmap_count <- amp_heatmap(amp_samp,
facet_by = "SampleCategory",
group_by = "species",
tax_aggregate = "Family",
tax_show = 40,
normalise = FALSE,
plot_values = FALSE,
plot_values_size = 3,
round = 0, color_vector = c("white", "#e5ba52", "#ab7ca3", "#9d02d7", "#0030bf"), plot_colorscale = "log10") + theme(axis.text.x = element_text(angle = 45, size=10, vjust = 1), axis.text.y = element_text(size=10), legend.position="right")
heatmap_count4.2. microbiomeutilities
Heatmap using microbiomeutilities
Create a detailed heatmap using the micro utilities package.
Subset taxa of interest
ps_tryp_otry = subset_taxa(ps_samp_tryp, Order=="Trypanosomatida")
ps_tryp_subtry = subset_taxa(ps_samp_tryp, Species=="Trypanosoma gilletti" | Species=="Trypanosoma sp. (cyclops-like)" | Species =="Trypanosoma vegrandis" | Species=="Trypanosoma noyesi" | Species=="Trypanosoma sp. (lewisi-like)" | Species =="Lotmaria passim")Creat plot
# create a gradient color palette for abundance
#grad_ab <- colorRampPalette(c("#faf3dd","#f7d486" ,"#5e6472"))
color_vector = colorRampPalette(c("#faf3dd", "#e5ba52", "#ab7ca3", "#9d02d7", "#0030bf"))
grad_ab_pal <- color_vector(10)
# create a color palette for varaibles of interest
meta_colors = list(c("Blood" = "#7a255d", "Tick" = "#9fd0cb", "Tissue" = "#7566ff"),
c("Brush tail possum" = "#440154FF", "Black rat" = "#482878FF", "Swamp rat"="#3E4A89FF", "Long-nosed bandicoot" ="#31688EFF", "Bush rat"="#26828EFF", "Brown antechinus" = "#1F9E89FF", "Rabbit"="#35B779FF", "Chuditch"= "#6DCD59FF", "Quenda" = "#B4DE2CFF", "Deer" = "#FDE725FF" ))
# add labels for pheatmap to detect
names(meta_colors) <- c("SampleCategory", "species")
ph_heatmap <- plot_taxa_heatmap(ps_tryp_subtry,
subset.top = 50,
VariableA = c("SampleCategory","species"),
heatcolors = grad_ab_pal, #rev(brewer.pal(6, "RdPu")),
transformation = "log10",
cluster_rows = T,
cluster_cols = F,
show_colnames = F,
annotation_colors=meta_colors, fontsize = 8)Top 50 OTUs selected log10, if zeros in data then log10(1+x) will be usedFirst top taxa were selected and
then abundances tranformed to log10(1+X)Warning in transform(phyobj1, "log10"): OTU table contains zeroes. Using log10(1
+ x) transform.ph_heatmap$plot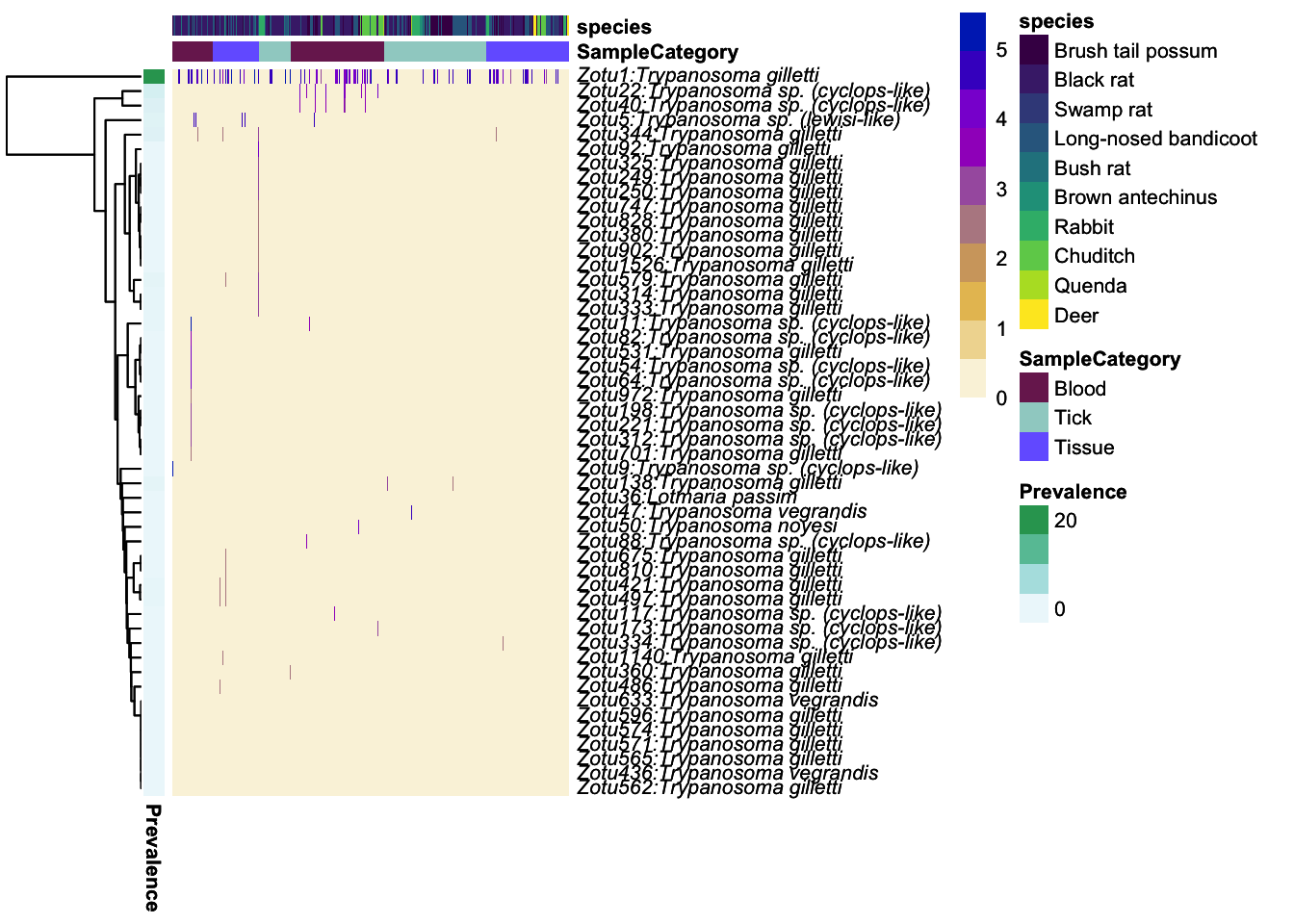
Save PDF of plots
ggsave("ph_heatmap.pdf", plot = ph_heatmap$plot, path = "output/plots/trypNGS", width = 25, height = 15, units = "cm")
sessionInfo()R version 4.0.3 (2020-10-10)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Mojave 10.14.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRblas.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
locale:
[1] en_AU.UTF-8/en_AU.UTF-8/en_AU.UTF-8/C/en_AU.UTF-8/en_AU.UTF-8
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] Biostrings_2.58.0 XVector_0.30.0
[3] patchwork_1.1.1 knitr_1.31
[5] microbiomeutilities_1.00.12 vegan_2.5-7
[7] lattice_0.20-41 permute_0.9-5
[9] DESeq2_1.30.1 SummarizedExperiment_1.20.0
[11] Biobase_2.50.0 MatrixGenerics_1.2.1
[13] matrixStats_0.58.0 GenomicRanges_1.42.0
[15] GenomeInfoDb_1.26.2 IRanges_2.24.1
[17] S4Vectors_0.28.1 BiocGenerics_0.36.0
[19] ape_5.4-1 data.table_1.14.0
[21] decontam_1.10.0 reshape_0.8.8
[23] microbiome_1.13.9 MicrobeR_0.3.2
[25] cowplot_1.1.1 viridis_0.5.1
[27] viridisLite_0.3.0 plotly_4.9.3
[29] agricolae_1.3-3 ggpubr_0.4.0
[31] ampvis2extras_0.1.5 ampvis2_2.6.7
[33] forcats_0.5.1 stringr_1.4.0
[35] dplyr_1.0.5 purrr_0.3.4
[37] readr_1.4.0 tidyr_1.1.2
[39] tibble_3.1.0 ggplot2_3.3.3
[41] tidyverse_1.3.0 phyloseq_1.34.0
[43] qiime2R_0.99.4 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] bit64_4.0.5 DelayedArray_0.16.2 rpart_4.1-15
[4] RCurl_1.98-1.2 doParallel_1.0.16 generics_0.1.0
[7] RSQLite_2.2.3 combinat_0.0-8 bit_4.0.4
[10] xml2_1.3.2 lubridate_1.7.10 httpuv_1.5.5
[13] assertthat_0.2.1 xfun_0.21 hms_1.0.0
[16] jquerylib_0.1.3 evaluate_0.14 promises_1.2.0.1
[19] fansi_0.4.2 progress_1.2.2 dbplyr_2.1.0
[22] readxl_1.3.1 igraph_1.2.6 DBI_1.1.1
[25] geneplotter_1.68.0 htmlwidgets_1.5.3 ellipsis_0.3.1
[28] crosstalk_1.1.1 backports_1.2.1 picante_1.8.2
[31] annotate_1.68.0 vctrs_0.3.7 abind_1.4-5
[34] cachem_1.0.4 withr_2.4.1 checkmate_2.0.0
[37] treeio_1.14.3 prettyunits_1.1.1 cluster_2.1.1
[40] lazyeval_0.2.2 crayon_1.4.1 genefilter_1.72.1
[43] labeling_0.4.2 pkgconfig_2.0.3 zCompositions_1.3.4
[46] nlme_3.1-152 nnet_7.3-15 rlang_0.4.10
[49] questionr_0.7.4 lifecycle_1.0.0 miniUI_0.1.1.1
[52] modelr_0.1.8 cellranger_1.1.0 rprojroot_2.0.2
[55] Matrix_1.3-2 aplot_0.0.6 phangorn_2.5.5
[58] carData_3.0-4 Rhdf5lib_1.12.1 reprex_1.0.0
[61] base64enc_0.1-3 pheatmap_1.0.12 whisker_0.4
[64] png_0.1-7 bitops_1.0-6 rhdf5filters_1.2.0
[67] blob_1.2.1 jpeg_0.1-8.1 rstatix_0.7.0
[70] DECIPHER_2.18.1 ggsignif_0.6.1 rtk_0.2.6.1
[73] klaR_0.6-15 scales_1.1.1 memoise_2.0.0
[76] magrittr_2.0.1 plyr_1.8.6 gghalves_0.1.1
[79] zlibbioc_1.36.0 compiler_4.0.3 philr_1.16.0
[82] RColorBrewer_1.1-2 cli_2.3.1 ade4_1.7-16
[85] htmlTable_2.1.0 Formula_1.2-4 MASS_7.3-53.1
[88] mgcv_1.8-34 tidyselect_1.1.0 stringi_1.5.3
[91] highr_0.8 yaml_2.2.1 locfit_1.5-9.4
[94] latticeExtra_0.6-29 ggrepel_0.9.1 grid_4.0.3
[97] sass_0.3.1 fastmatch_1.1-0 tools_4.0.3
[100] rio_0.5.26 rstudioapi_0.13 foreach_1.5.1
[103] foreign_0.8-81 git2r_0.28.0 gridExtra_2.3
[106] farver_2.0.3 Rtsne_0.15 digest_0.6.27
[109] rvcheck_0.1.8 BiocManager_1.30.10 shiny_1.6.0
[112] quadprog_1.5-8 Rcpp_1.0.6 car_3.0-10
[115] broom_0.7.5 later_1.1.0.1 httr_1.4.2
[118] AnnotationDbi_1.52.0 colorspace_2.0-0 rvest_0.3.6
[121] XML_3.99-0.5 fs_1.5.0 truncnorm_1.0-8
[124] splines_4.0.3 tidytree_0.3.3 multtest_2.46.0
[127] xtable_1.8-4 jsonlite_1.7.2 ggtree_2.4.1
[130] AlgDesign_1.2.0 R6_2.5.0 Hmisc_4.5-0
[133] NADA_1.6-1.1 pillar_1.5.1 htmltools_0.5.1.1
[136] mime_0.10 glue_1.4.2 fastmap_1.1.0
[139] DT_0.17 BiocParallel_1.24.1 ggnet_0.1.0
[142] codetools_0.2-18 utf8_1.2.1 bslib_0.2.4.9001
[145] network_1.16.1 curl_4.3 zip_2.1.1
[148] openxlsx_4.2.3 survival_3.2-7 rmarkdown_2.7.2
[151] biomformat_1.18.0 munsell_0.5.0 rhdf5_2.34.0
[154] GenomeInfoDbData_1.2.4 iterators_1.0.13 labelled_2.7.0
[157] haven_2.3.1 reshape2_1.4.4 gtable_0.3.0