Analyses
Tina Lasisi
2023-03-06 11:51:46
Last updated: 2023-03-06
Checks: 6 1
Knit directory: PODFRIDGE/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230302) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 5f805fe. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rproj.user/
Ignored: data/.DS_Store
Unstaged changes:
Modified: analysis/analyses.Rmd
Modified: analysis/methods.Rmd
Modified: data/us-pop-detailed-multisheet.xls
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/analyses.Rmd) and HTML
(docs/analyses.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 5f805fe | Tina Lasisi | 2023-03-05 | Build site. |
| Rmd | 5106bab | Tina Lasisi | 2023-03-05 | add analyses |
| html | c3948af | Tina Lasisi | 2023-03-04 | Build site. |
| html | f02bc38 | Tina Lasisi | 2023-03-02 | Build site. |
| html | c9130d5 | Tina Lasisi | 2023-03-02 | wflow_git_commit(all = TRUE) |
| html | a4a7d45 | Tina Lasisi | 2023-03-02 | Build site. |
| html | 00073fd | Tina Lasisi | 2023-03-02 | Build site. |
| html | 51ed5a6 | Tina Lasisi | 2023-03-02 | Build site. |
| Rmd | 13ed9ae | Tina Lasisi | 2023-03-02 | Publishing POPFORGE |
| html | 13ed9ae | Tina Lasisi | 2023-03-02 | Publishing POPFORGE |
# Load necessary packages
library(wesanderson) # for color palettes
library(tidyverse) #data wrangling etc── Attaching packages ─────────────────────────────────────── tidyverse 1.3.2 ──
✔ ggplot2 3.4.0 ✔ purrr 0.3.5
✔ tibble 3.1.8 ✔ dplyr 1.0.10
✔ tidyr 1.2.1 ✔ stringr 1.5.0
✔ readr 2.1.3 ✔ forcats 0.5.2
── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()# Set path to the data file
path <- "./data/"
savepath <- "./output/"
# Set up vector for cousin degree
p <- c(1:8)
# Set up initial population size
N <- 76e6
# Read in data on US population sizes by year
US_pop <- read.csv(paste(path, "est-pop-combo.csv", sep = ""))
# Calculate number of grandparents by generation
p_grandpar_gen <- 1990 - 30 * (p + 1)
# Get population sizes by year for grandparents' generation
US_Ns <- US_pop %>%
filter(Year %in% p_grandpar_gen)
# Scale population size down by 50% (assumed number of potential parents) and 90% of those have children + set minimum for populations
# US_Ns %>%
# mutate(across(!Year, ~ . * 0.5 * 0.9))
N <- US_Ns %>%
mutate(across(!Year, ~ case_when(. * 0.5 * 0.9 < 1e6 ~ 1e6,
TRUE ~ . * 0.5 * 0.9)))
# Set up vector of database sizes to test
DB.sizes <- c(1e6, 5e6, 10e6)
DB.sizes.EUR <- DB.sizes*0.8
DB.sizes.AFR <- DB.sizes*0.05
# Set color palette for graphs
my.cols <- wes_palette("Darjeeling1")N Year Total White Black
1 1720 1000000 1000000 1000000
2 1750 1000000 1000000 1000000
3 1780 1251166 1000000 1000000
4 1810 3257946 2637933 1000000
5 1840 7678509 6385367 1293142
6 1870 17351267 15115220 2196004
7 1900 34197559 30064138 3975297
8 1930 55248771 49629033 5351014Probability of p-th degree cousin
plot_prob_DB_size <- function(p, DB.sizes, N, my.cols) {
plot(c(1, 8), c(0, 1), type = "n", ylab = "Probability of at least one p-th cousin in database", xlab = "p (degree of cousin)")
sapply(1:length(DB.sizes), function(i) {
DB.size <- DB.sizes[i]
prob.no.rellys <- exp(-2^(2*p - 2) * DB.size / N)
points(p, 1 - prob.no.rellys, type = "b", col = my.cols[i], pch = 19, lwd = 1.5)
})
legend(x = "bottomright", legend = c("Database size (Millions) =", format(DB.sizes / 1e6, dig = 1)), col = c(NA, my.cols), pch = 19)
}
layout(t(1:2))
# plot_prob_DB_size(p, DB.sizes.EUR, N$Total, my.cols)
plot_prob_DB_size(p, DB.sizes.EUR, N$White, my.cols)
# plot_prob_DB_size(p, DB.sizes.EUR, N$White, my.cols)
# plot_prob_DB_size(p, DB.sizes.EUR, N$Total, my.cols)
plot_prob_DB_size(p, DB.sizes.AFR, N$Black, my.cols)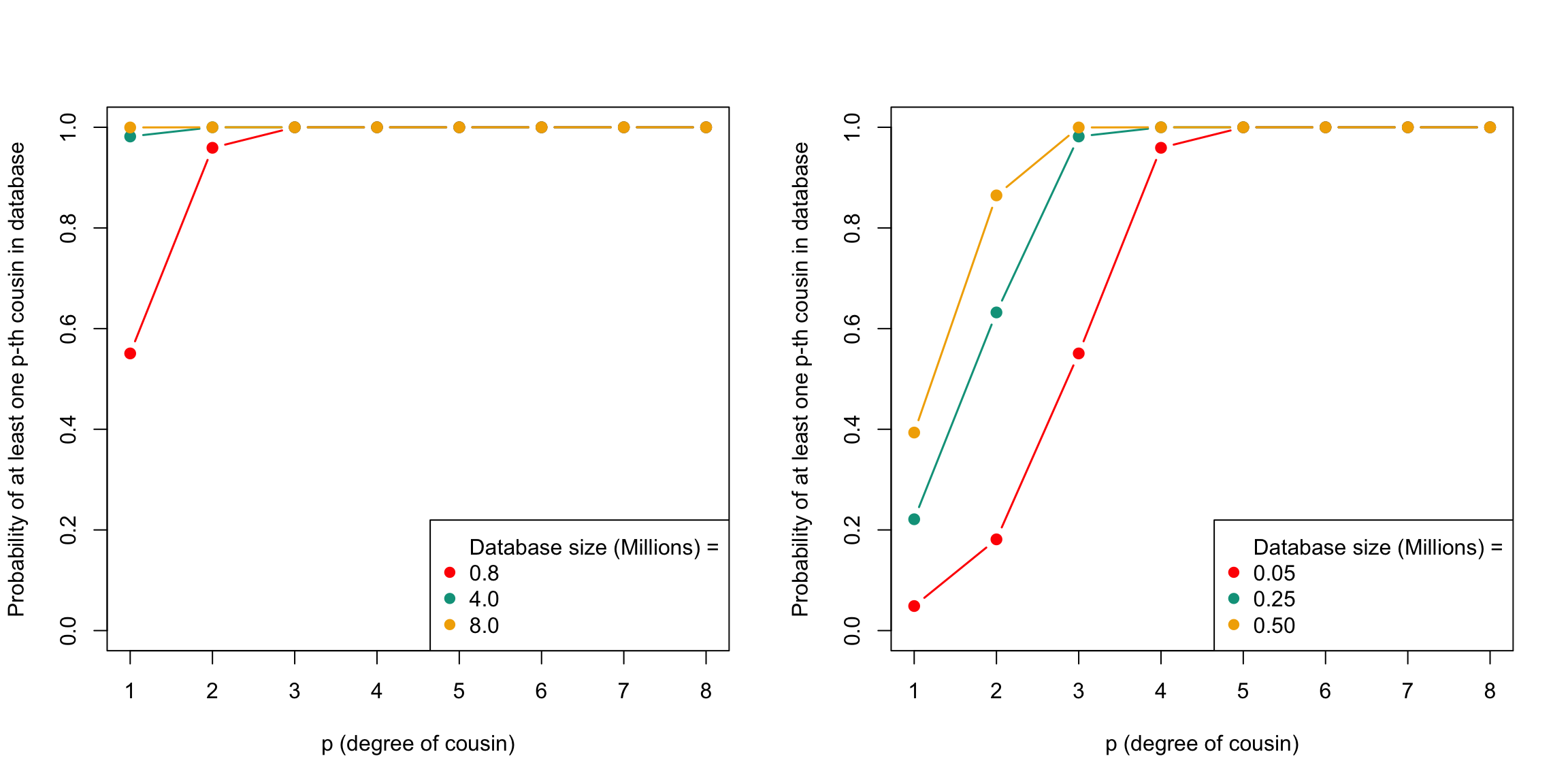
The plot above displays the probability of having at least one p-th cousin in a genetic database and the expected number of p-th cousins in a database. The plot shows the probability of finding at least one p-th cousin in a database of a given size for White Americans (left) and Black Americans (right)
| Version | Author | Date |
|---|---|---|
| 5f805fe | Tina Lasisi | 2023-03-05 |
# plot_prob_DB_size(p, DB.sizes.AFR, N$Total, my.cols)Number of p-th degree cousins
# Plot of expected number of p-th cousins in sample
expected_cousins <- function(p, DB.sizes, N, my.cols) {
plot(c(1,8), c(0,1000), type = "n", ylab = "Expected number of p-th cousins in database", xlab = "p.(degree of cousin)")
sapply(1:length(DB.sizes), function(i){
num_cousins <- 4^(p) * DB.sizes[i] / (N / 2)
points(p, num_cousins, type = "b", col = my.cols[i], lwd = 1.5, pch = 19)
})
}
layout(t(1:2))
expected_cousins(p, DB.sizes.EUR, N$White, my.cols)[[1]]
NULL
[[2]]
NULL
[[3]]
NULLexpected_cousins(p, DB.sizes.AFR, N$Black, my.cols)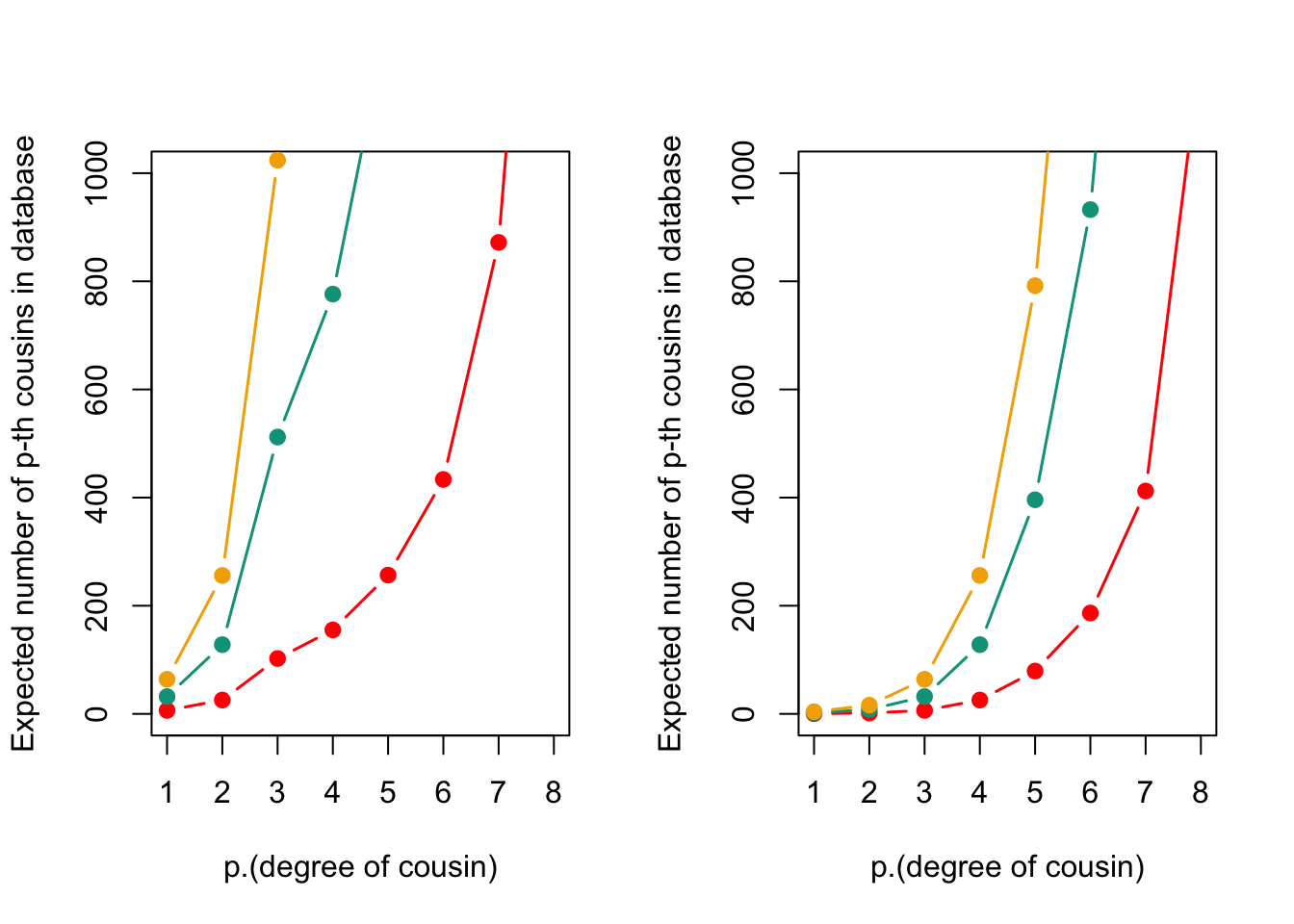
This figure the expected number of cousins for White Americans (left) and Black Americans (right) based on database sizes of 1 million, 5 million, and 10 million individuals. The expected number of cousins increases with increasing database size and degree of relatedness
| Version | Author | Date |
|---|---|---|
| 5f805fe | Tina Lasisi | 2023-03-05 |
[[1]]
NULL
[[2]]
NULL
[[3]]
NULLProbability of a genetically detectable cousin
Below, we calculate the expected number of shared blocks of genetic material between cousins of varying degrees of relatedness. This is important because the probability of detecting genetic material that is shared between two individuals decreases as the degree of relatedness between them decreases. The code uses a Poisson distribution assumption to estimate the probability of two cousins sharing at least one, two, or three blocks of genetic material, based on the expected number of shared blocks of genetic material calculated from previous research.
# The variable 'meiosis' represents the number of meiosis events between cousins, where 'p' is the degree of relatedness (i.e. p = 1 for first cousins, p = 2 for second cousins, etc.)
meiosis <- p + 1
## Expected number of blocks shared between cousins
# 'E.num.blocks' is the expected number of blocks of shared genetic material between cousins based on the degree of relatedness and the number of meiosis events between them. This value is calculated based on previous research and is not calculated in this code.
E.num.blocks <- 2 * (33.8 * (2 * meiosis) + 22) / (2^(2 * meiosis - 1))
## Use Poisson assumption
# 'Prob.genetic' is the probability of two cousins sharing at least one block of genetic material based on the expected number of shared blocks calculated in the previous step. The calculation uses a Poisson distribution assumption.
Prob.genetic <- 1 - exp(-E.num.blocks)
# 'prob.g.e.2.blocks' is the probability of two cousins sharing at least two blocks of genetic material based on the expected number of shared blocks calculated in the previous step. The calculation uses a Poisson distribution assumption.
prob.g.e.2.blocks <- 1 - sapply(E.num.blocks, function(expected.num) {sum(dpois(0:1, expected.num))})
# 'prob.g.e.3.blocks' is the probability of two cousins sharing at least three blocks of genetic material based on the expected number of shared blocks calculated in the previous step. The calculation uses a Poisson distribution assumption.
prob.g.e.3.blocks <- 1 - sapply(E.num.blocks, function(expected.num) {sum(dpois(0:2, expected.num))})General
## Plot for number of shared blocks with p-th degree cousins
# Set layout of plot
layout(t(1))
# Set color palette for plot
my.cols2<-wes_palette("FantasticFox1")[3:5]
# Create a blank plot with labeled axes
plot(c(1,8),c(0,1),type="n",ylab="Probability p-th cousin \"detectable\"",xlab="p.(degree of cousin)")
# Add points for probability of detecting pth cousin with genomic blocks using colors from my.cols2
points(p,Prob.genetic,col=my.cols2[1],pch=19,type="b",lwd=2)
points(p,prob.g.e.2.blocks,col=my.cols2[2],pch=19,type="b",lwd=2)
points(p,prob.g.e.3.blocks,col=my.cols2[3],pch=19,type="b",lwd=2)
# Add a legend to the plot
legend(x="topright",legend=c("Cousins (w. >0 genomic blocks)","Cousins (w. >1 genomic blocks)","Cousins (w. >2 genomic blocks)"),col=my.cols2[1:3],lty=1)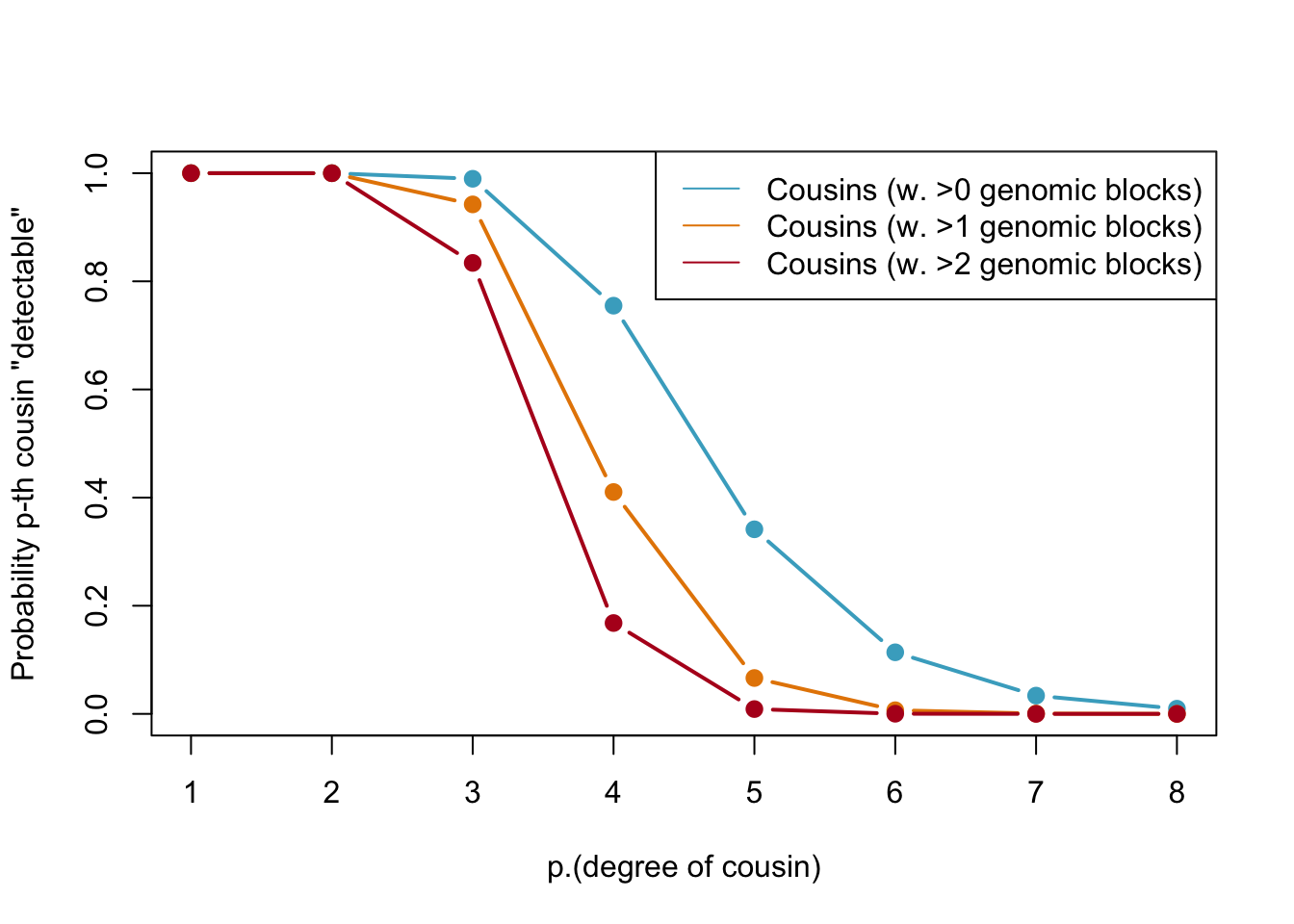
Probabilities of detecting a genetic cousin in a database based on shared genomic blocks. Blue lines represent cousins with at least one genomic block, orange dotted and red lines represent cousins with at least two and three genomic blocks, respectively. The legend specifies the type of cousin being represented by each line.
| Version | Author | Date |
|---|---|---|
| 5f805fe | Tina Lasisi | 2023-03-05 |
Relative to database size
plt_cousins_database <- function(p, DB.sizes, N, prob.g.e.3.blocks, my.cols) {
# Create plot
plot(c(1, 8), c(0, 500), type = "n", ylab = "Expected number of genetic p-th cousins in database", xlab = "p. (degree of cousin)")
# Calculate and plot expected number of genetic cousins for each database size
sapply(1:length(DB.sizes), function(i) {
num.cousins <- 4^(p) * DB.sizes[i] / (N / 2)
points(p, num.cousins * prob.g.e.3.blocks, type = "b", lty = 1, col = my.cols[i])
})
# Create legend for plot
my.leg <- c(c("Genetic Cousins", ">2 blocks"), c("Database size (Millions) =", format(DB.sizes / 1e6, dig = 1)))
legend(x = "topright", legend = my.leg, col = c(NA, rep("black", 1), NA, my.cols), lty = c(NA, 1:2, rep(NA, 3)), pch = c(rep(NA, 3), rep(19, 3)))
}
layout(t(1:2))
# plt_cousins_database(p, DB.sizes, N$White, prob.g.e.3.blocks, my.cols)
plt_cousins_database(p, DB.sizes.EUR, N$White, prob.g.e.3.blocks, my.cols)
plt_cousins_database(p, DB.sizes.AFR, N$Black, prob.g.e.3.blocks, my.cols)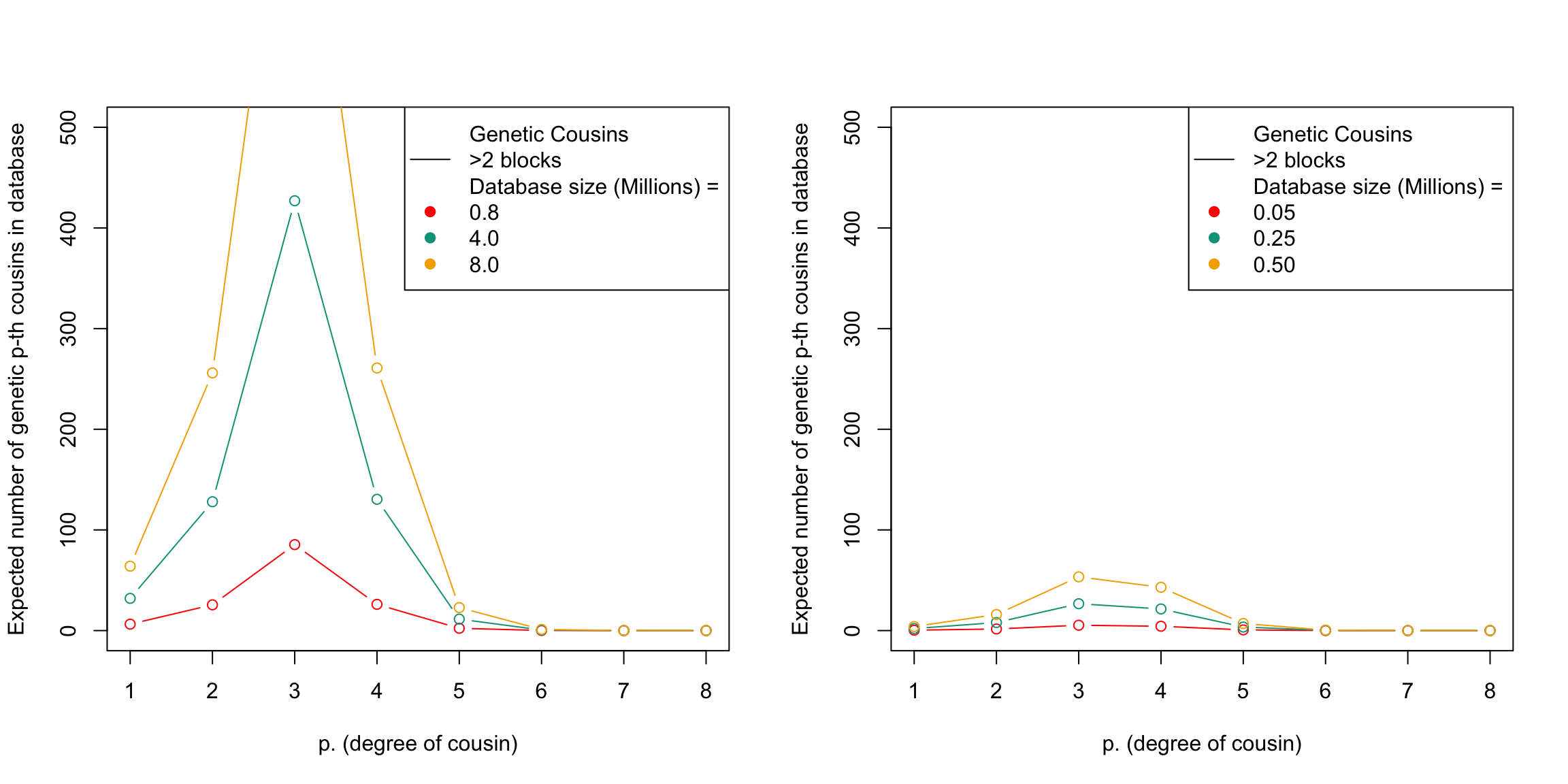
This figure shows the expected number of genetic relatives of degree p in a database of a given size. The x-axis represents the degree of relatedness
| Version | Author | Date |
|---|---|---|
| 5f805fe | Tina Lasisi | 2023-03-05 |
sessionInfo()R version 4.2.2 (2022-10-31)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Ventura 13.0.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2-arm64/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] forcats_0.5.2 stringr_1.5.0 dplyr_1.0.10 purrr_0.3.5
[5] readr_2.1.3 tidyr_1.2.1 tibble_3.1.8 ggplot2_3.4.0
[9] tidyverse_1.3.2 wesanderson_0.3.6 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.9 lubridate_1.9.0 getPass_0.2-2
[4] ps_1.7.2 assertthat_0.2.1 rprojroot_2.0.3
[7] digest_0.6.30 utf8_1.2.2 R6_2.5.1
[10] cellranger_1.1.0 backports_1.4.1 reprex_2.0.2
[13] evaluate_0.18 highr_0.9 httr_1.4.4
[16] pillar_1.8.1 rlang_1.0.6 readxl_1.4.1
[19] googlesheets4_1.0.1 rstudioapi_0.14 whisker_0.4
[22] callr_3.7.3 jquerylib_0.1.4 rmarkdown_2.18
[25] googledrive_2.0.0 munsell_0.5.0 broom_1.0.1
[28] modelr_0.1.10 compiler_4.2.2 httpuv_1.6.6
[31] xfun_0.35 pkgconfig_2.0.3 htmltools_0.5.3
[34] tidyselect_1.2.0 fansi_1.0.3 crayon_1.5.2
[37] withr_2.5.0 tzdb_0.3.0 dbplyr_2.2.1
[40] later_1.3.0 grid_4.2.2 jsonlite_1.8.3
[43] gtable_0.3.1 lifecycle_1.0.3 DBI_1.1.3
[46] git2r_0.30.1 magrittr_2.0.3 scales_1.2.1
[49] cli_3.4.1 stringi_1.7.8 cachem_1.0.6
[52] fs_1.5.2 promises_1.2.0.1 xml2_1.3.3
[55] bslib_0.4.1 ellipsis_0.3.2 generics_0.1.3
[58] vctrs_0.5.1 tools_4.2.2 glue_1.6.2
[61] hms_1.1.2 processx_3.8.0 fastmap_1.1.0
[64] yaml_2.3.6 timechange_0.1.1 colorspace_2.0-3
[67] gargle_1.2.1 rvest_1.0.3 knitr_1.41
[70] haven_2.5.1 sass_0.4.4