Sexual selection and the population genetics of a selfish gene
Supplementary methods
Thomas Keaney, Theresa Jones and Luke Holman
Last updated: 2021-06-02
Checks: 7 0
Knit directory: SD_sexual_selection/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200925) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 0289923. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rapp.history
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.Rapp.history
Unstaged changes:
Modified: SD_ESM_references.bib
Modified: SD_crossing_scheme.png
Modified: SD_sexual_selection.Rproj
Modified: Supplementary_material.Rmd
Modified: analysis/Population_genetic_model.Rmd
Modified: analysis/_site.yml
Modified: data/Drive_test.csv
Modified: data/SD_mating_data.csv
Modified: data/two_choice_test_data.csv
Modified: fits/remating_model_attractiveness.rds
Modified: fits/survival_model_censored_attract.rds
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/Supp_methods.Rmd) and HTML (docs/Supp_methods.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 0289923 | tomkeaney | 2021-06-02 | Edits to make the manuscript smaller |
library(png) # to load images
library(grid) # to plot images
library(tidyverse) # for data wrangling and plotting-- Attaching packages --------------------------------------- tidyverse 1.3.0 --v ggplot2 3.3.3 v purrr 0.3.4
v tibble 3.1.0 v dplyr 1.0.5
v tidyr 1.1.3 v stringr 1.4.0
v readr 1.4.0 v forcats 0.5.1Warning: package 'ggplot2' was built under R version 4.0.3Warning: package 'tibble' was built under R version 4.0.4Warning: package 'tidyr' was built under R version 4.0.4Warning: package 'dplyr' was built under R version 4.0.4Warning: package 'forcats' was built under R version 4.0.3-- Conflicts ------------------------------------------ tidyverse_conflicts() --
x dplyr::filter() masks stats::filter()
x dplyr::lag() masks stats::lag()library(pander) # for tablesPilot experiment: confirming that SD exhibits segregation distortion
In a pilot experiment, we measured the strength of segregation distortion produced by each of our experimental treatment lines. We crossed females from each of the three SD/+ lines and the +/+ line to males homozygous for the bw mutation (Figure S1); like SD, bw is located on chromosome 2, so this cross yielded SD/bw or +/bw progeny. We then mated 20 SD/bw (or +/bw) males from each of the four crosses to bw/bw females, and recorded the eye colour (red or brown) of the resulting female offspring to determine the proportion of offspring fertilised by SD- (or +) and bw-bearing sperm. Male progeny were not counted because some of them in the reciprocal cross (see below) expressed a white-eye phenotype (due to male hemizygosity and an X-linked mutation of white), preventing us from determining which copy of chromosome 2 they inherited.
SD alleles are commonly associated with viability costs, which might cause underestimation of the strength of segregation distortion. To correct for any such viability costs, we also performed the reciprocal cross ( SD/+ females × bw/bw males) and calculated the proportion of offspring inheriting the SD bearing chromosome as above. Because SD does not affect segregation in females, a shortage of adult offspring carrying SD (relative to the 50% Mendelian expectation) indicates reduced survival of SD progeny to adulthood (relative to bw progeny). We calculated the viability-corrected estimate of segregation distortion, kc, using the formula in Temin [1].
To analyse our results, we fit a binomial model, in which red-eye daughters (i.e. the progeny that inherited the SD or + allele from their SD/bw or +/bw father) were treated as ‘successes’ and the brown-eye daughters as ‘failures’. We included the sex of the experimental individual and the variant of SD (or control) as fixed effects (with the control as the reference level), as well as the interaction between these variables. We also included pair ID as a random effect.
\(~\)
Table S1: Recipe for food medium used in our experiment. The provided quantities make ~ 1 litre of food.
tibble("Ingredients" = c("Soy flour", "Cornmeal", "Yeast", "Dextrose", "Agar", "Water", "Tegosept", "Acid mix (4 mL orthophosphoric acid, 41 mL propionic acid, 55 mL water to make 100 mL)"),
"Quantity" = c("20 g", "73 g", "35 g", "75 g", "6 g", "1000 mL", "17 mL", "14 mL")) %>%
pander(split.cell = 40, split.table = Inf)| Ingredients | Quantity |
|---|---|
| Soy flour | 20 g |
| Cornmeal | 73 g |
| Yeast | 35 g |
| Dextrose | 75 g |
| Agar | 6 g |
| Water | 1000 mL |
| Tegosept | 17 mL |
| Acid mix (4 mL orthophosphoric acid, 41 mL propionic acid, 55 mL water to make 100 mL) | 14 mL |
\(~\)
img <- readPNG("SD_crossing_scheme.png")
grid.raster(img)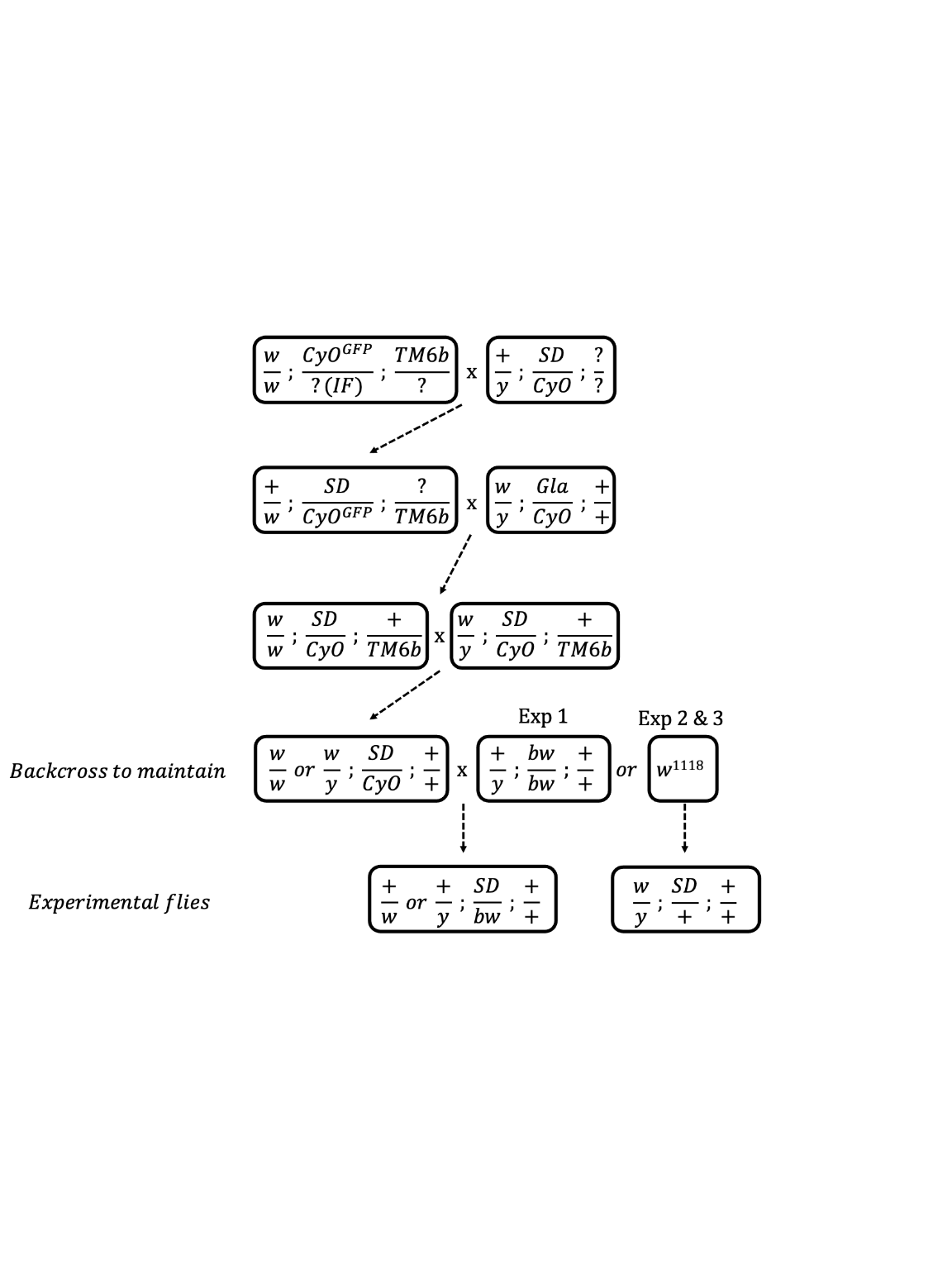
Figure S1. Crossing scheme used to standardise the genetic background across the SD-5/+, SD-72/+, SD-Mad/+ and SD+/+ lines. The SD+/+ line was created in identical fashion except that we substituted the SD bearing chromosome with chromosome 2 from the w1118 isogenic line. Note that at step four there are three possible options 1) the leftmost genotype can be backcrossed to maintain it in the laboratory, 2) the leftmost genotype can be crossed to a bw stock to produce the experimental flies used in Experiment 1 or 3) the leftmost genotype can be crossed to w1118 to create the experimental flies used in Experiments 2 and 3.
sessionInfo()R version 4.0.2 (2020-06-22)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19041)
Matrix products: default
locale:
[1] LC_COLLATE=English_United Kingdom.1252
[2] LC_CTYPE=English_United Kingdom.1252
[3] LC_MONETARY=English_United Kingdom.1252
[4] LC_NUMERIC=C
[5] LC_TIME=English_United Kingdom.1252
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] pander_0.6.3 forcats_0.5.1 stringr_1.4.0 dplyr_1.0.5
[5] purrr_0.3.4 readr_1.4.0 tidyr_1.1.3 tibble_3.1.0
[9] ggplot2_3.3.3 tidyverse_1.3.0 png_0.1-7 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] Rcpp_1.0.6 lubridate_1.7.10 ps_1.6.0 assertthat_0.2.1
[5] rprojroot_2.0.2 digest_0.6.27 utf8_1.2.1 R6_2.5.0
[9] cellranger_1.1.0 backports_1.2.1 reprex_1.0.0 evaluate_0.14
[13] highr_0.8 httr_1.4.2 pillar_1.5.1 rlang_0.4.10
[17] readxl_1.3.1 rstudioapi_0.13 whisker_0.4 jquerylib_0.1.3
[21] rmarkdown_2.7 munsell_0.5.0 broom_0.7.5 compiler_4.0.2
[25] httpuv_1.5.5 modelr_0.1.8 xfun_0.22 pkgconfig_2.0.3
[29] htmltools_0.5.1.1 tidyselect_1.1.0 fansi_0.4.2 crayon_1.4.1
[33] dbplyr_2.1.0 withr_2.4.1 later_1.1.0.1 jsonlite_1.7.2
[37] gtable_0.3.0 lifecycle_1.0.0 DBI_1.1.1 git2r_0.28.0
[41] magrittr_2.0.1 scales_1.1.1 cli_2.3.1 stringi_1.5.3
[45] fs_1.5.0 promises_1.2.0.1 xml2_1.3.2 bslib_0.2.4
[49] ellipsis_0.3.1 generics_0.1.0 vctrs_0.3.6 tools_4.0.2
[53] glue_1.4.2 hms_1.0.0 yaml_2.2.1 colorspace_2.0-0
[57] rvest_1.0.0 knitr_1.31 haven_2.3.1 sass_0.3.1