UMAP on 7 MOFA factors
## UMAP
plot.umap.data <- plot_dimred(mofa, method="UMAP", color_by = "condition",stroke = 0.001, dot_size =1, alpha = 0.2, return_data = T)
plot.umap.all <- ggplot(plot.umap.data, aes(x=x, y = y, fill = color_by))+
geom_point(size = 0.8, alpha = 0.6, shape = 21, stroke = 0) +
theme_half_open() +
scale_fill_manual(values = colorgradient6_manual, labels = c(labels), name = "Time aIg",)+
theme(legend.position="none")+
add.textsize +
labs(title = "UMAP on MOFA+ factors shows minute and \nhour time-scale cell-state transitions", x = "UMAP 1", y = "UMAP 2")
## UMAP legend
legend.umap <- get_legend( ggplot(plot.umap.data, aes(x=x, y = y, fill = color_by))+
geom_point(size = 2, alpha = 1, shape = 21, stroke = 0) +
theme_half_open() +
scale_fill_manual(values = colorgradient6_manual, labels = c(labels), name = "Time aIg \n(minutes)",)+
add.textsize +
labs(title = "UMAP on MOFA+ factors shows minute and \nhour time-scale cell-state transitions", x = "UMAP 1", "y = UMAP 2"))
legend.umap <- as_ggplot(legend.umap)
plot_fig1_umap <- plot_grid(plot.umap.all,legend.umap, labels = c(""), label_size = 10, ncol = 2, rel_widths = c(1, 0.2))
ggsave(plot_fig1_umap, filename = "output/paper_figures/Fig1_UMAP.pdf", width = 68, height = 62, units = "mm", dpi = 300, useDingbats = FALSE)
ggsave(plot_fig1_umap, filename = "output/paper_figures/Fig1_UMAP.png", width = 68, height = 62, units = "mm", dpi = 300)
plot_fig1_umap
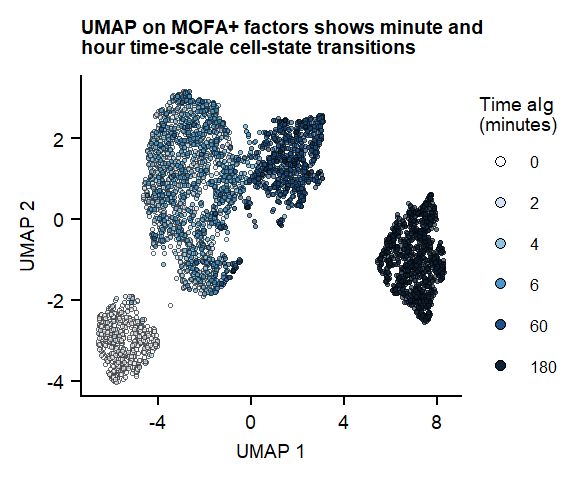 Figure 1. UMAP of 7 MOFA factors, integrating phospho-protein and RNA measurements
Figure 1. UMAP of 7 MOFA factors, integrating phospho-protein and RNA measurements
Suppl PCA
seu_combined_selectsamples <- RunPCA(seu_combined_selectsamples,assay = "SCT.RNA", features = genes.variable, verbose = FALSE, ndims.print = 0, reduction.name = "pca.RNA")
seu_combined_selectsamples <- RunPCA(seu_combined_selectsamples, assay = "PROT", features = proteins.all, verbose = FALSE, ndims.print = 0,reduction.name = "pca.PROT")
## PCA analysis
plot.PCA.RNA <- DimPlot(seu_combined_selectsamples, reduction = "pca.RNA", group.by = "condition", pt.size =0.2) +
scale_color_manual(values = colorgradient6_manual2, labels = c(labels), name = "Time aIg",)+
labs(title = "RNA PCA separates cells \nat hour time-scale", x= "RNA PC 1", y = " RNA PC 2") +
add.textsize +
theme(legend.position = "none")
plot.PCA.PROT <- DimPlot(seu_combined_selectsamples, reduction = "pca.PROT", group.by = "condition", pt.size = 0.2) +
scale_color_manual(values = colorgradient6_manual2, labels = c(labels), name = "Time aIg",)+
labs(title = "(phospho-)protein PCA separates cells\nat minutes time-scale", x= "Protein PC 1", y = "Protein PC 2") +
add.textsize +
# scale_x_reverse()+
theme(legend.position = "none")
legend <- get_legend(DimPlot(seu_combined_selectsamples, reduction = "pca.RNA", group.by = "condition", pt.size =0.1) +
scale_color_manual(values = colorgradient6_manual2, labels = c(labels), name = "Time aIg",)+
add.textsize)
legend <- as_ggplot(legend)
PCA.PROTPC1.data <- data.frame(rank = 1:80,
protein = names(sort(seu_combined_selectsamples@reductions$pca.PROT@feature.loadings[,1])),
weight.PC1 = sort(seu_combined_selectsamples@reductions$pca.PROT@feature.loadings[,1]),
highlights = c(names(sort(seu_combined_selectsamples@reductions$pca.PROT@feature.loadings[,1]))[1:4],rep("",76))
)
plot.PCA.PROTweightsPC1 <- ggplot(PCA.PROTPC1.data, aes(x=weight.PC1, y = rank, label = highlights)) +
geom_point(size=0.1) +
labs(title = "Top 4 protein loadings \n", x= "Weight Protein PC1") +
geom_point(color = ifelse(PCA.PROTPC1.data$highlights == "", "grey50", "red")) +
geom_text_repel(size = 2, segment.size = 0.25)+
theme_half_open()+
add.textsize +
theme(axis.title.y=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank()
) +
scale_x_reverse()+
scale_y_reverse()
PCA.RNAPC1.data <- data.frame(rank = 1:2371,
RNA = names(sort(seu_combined_selectsamples@reductions$pca.RNA@feature.loadings[,1])),
weight.PC1 = sort(seu_combined_selectsamples@reductions$pca.RNA@feature.loadings[,1]),
highlights = c(rep("",2366),names(sort(seu_combined_selectsamples@reductions$pca.RNA@feature.loadings[,1]))[2367:2371])
)
## Loadings RNA
plot.PCA.RNAweightsPC1 <- ggplot(PCA.RNAPC1.data, aes(x=weight.PC1, y = rank, label = highlights)) +
geom_point(size=0.1) +
labs(title = "Top 4 RNA loadings \n", x= "Weight RNA PC1") +
geom_point(color = ifelse(PCA.RNAPC1.data$highlights == "", "grey50", "red")) +
geom_text_repel(size = 2, segment.size = 0.25)+
theme_half_open()+
add.textsize +
theme(axis.title.y=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)
## ridgeplots
PC1.data <- data.frame(sample = rownames(seu_combined_selectsamples@reductions$pca.PROT@cell.embeddings),
PC1_PROT = seu_combined_selectsamples@reductions$pca.PROT@cell.embeddings[,1],
PC1_RNA = seu_combined_selectsamples@reductions$pca.RNA@cell.embeddings[,1]) %>%
left_join(meta.allcells)
plot_ridge_PC1Prot <- ggplot(PC1.data, aes(x = PC1_PROT, y = condition, fill = condition)) +
scale_fill_manual(values = colorgradient6_manual, labels = c(labels), name = "Time aIg \n(minutes)")+
geom_density_ridges2() +
scale_y_discrete(labels = labels, name = "Time aIg (minutes)")+
scale_x_continuous(name = "PC1 Proteins") +
theme_half_open() +
add.textsize+
theme(legend.position = "none")
plot_ridge_PC1RNA <- ggplot(PC1.data, aes(x = PC1_RNA, y = condition, fill = condition)) +
scale_fill_manual(values = colorgradient6_manual, labels = c(labels), name = "Time aIg \n(minutes)")+
geom_density_ridges2() +
scale_y_discrete(labels = labels, name = "Time aIg (minutes)")+
scale_x_continuous(name = "PC1 RNA") +
theme_half_open() +
add.textsize+
theme(legend.position = "none")
Fig1.pca <- plot_grid(plot.PCA.PROT,plot.PCA.RNA,legend, plot_ridge_PC1Prot, plot_ridge_PC1RNA,NULL, plot.PCA.PROTweightsPC1, plot.PCA.RNAweightsPC1, labels = c(panellabels[1:2], "","","","",panellabels[3:4]), label_size = 10, ncol = 3, rel_widths = c(0.9,0.9,0.2,0.9,0.9), rel_heights = c(1,0.8,1.3))
Picking joint bandwidth of 0.37
Picking joint bandwidth of 0.368
ggsave(filename = "output/paper_figures/Suppl_PCA_aIg.pdf", width = 143, height = 170, units = "mm", dpi = 300, useDingbats = FALSE)
ggsave(filename = "output/paper_figures/Suppl_PCA_aIg.png", width = 143, height = 170, units = "mm", dpi = 300)
Fig1.pca
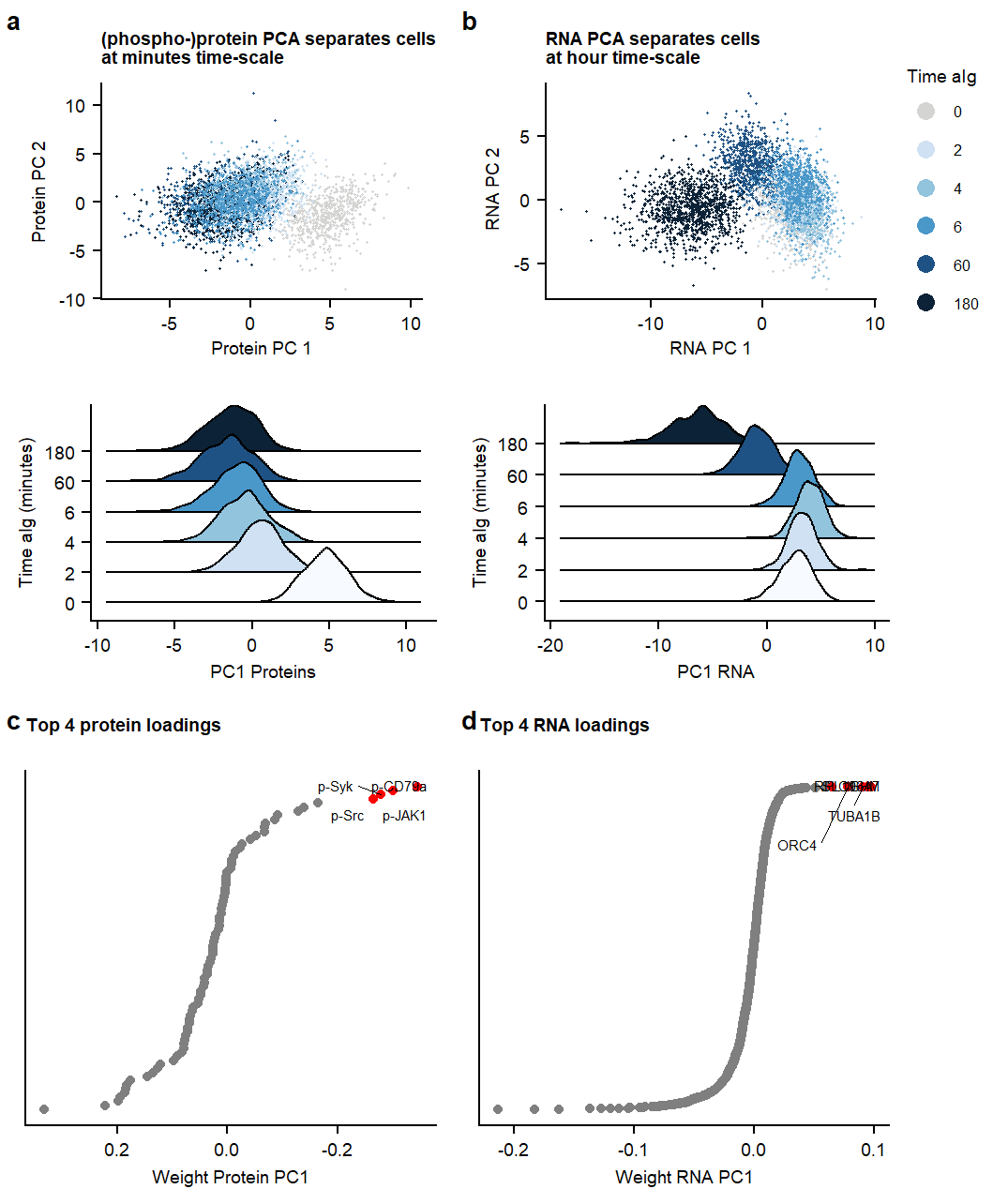 Supplemental Figure. PCA analysis on RNA ór Protein dataset
Supplemental Figure. PCA analysis on RNA ór Protein dataset
Suppl MOFA model properties
Fig.1.suppl.mofa.row1 <- plot_grid(plot.variance.perfactor.all, plot.variance.total,NULL, plot.heatmap.pval.covariates, labels = c(panellabels[1:3]), label_size = 10, ncol = 4, rel_widths = c(1.35, 0.27,0.2,0.38))
Fig.1.suppl.mofa.row2 <- plot_grid(plot.violin.factorall,legend.umap, labels = panellabels[4], label_size = 10, ncol = 2, rel_widths = c(1,0.1))
Suppl_mofa <- plot_grid(Fig.1.suppl.mofa.row1, Fig.1.suppl.mofa.row2, plot.rank.PROT.2.4to7, plot.rank.RNA.2.4to7, labels = c("","", panellabels[5:6]),label_size = 10, ncol = 1, rel_heights = c(1.45,1,1.1,1.1))
ggsave(Suppl_mofa, filename = "output/paper_figures/Suppl_MOFAaIg.pdf", width = 183, height = 220, units = "mm", dpi = 300, useDingbats = FALSE)
ggsave(Suppl_mofa, filename = "output/paper_figures/Suppl_MOFAaIg.png", width = 183, height = 220, units = "mm", dpi = 300)
Suppl_mofa
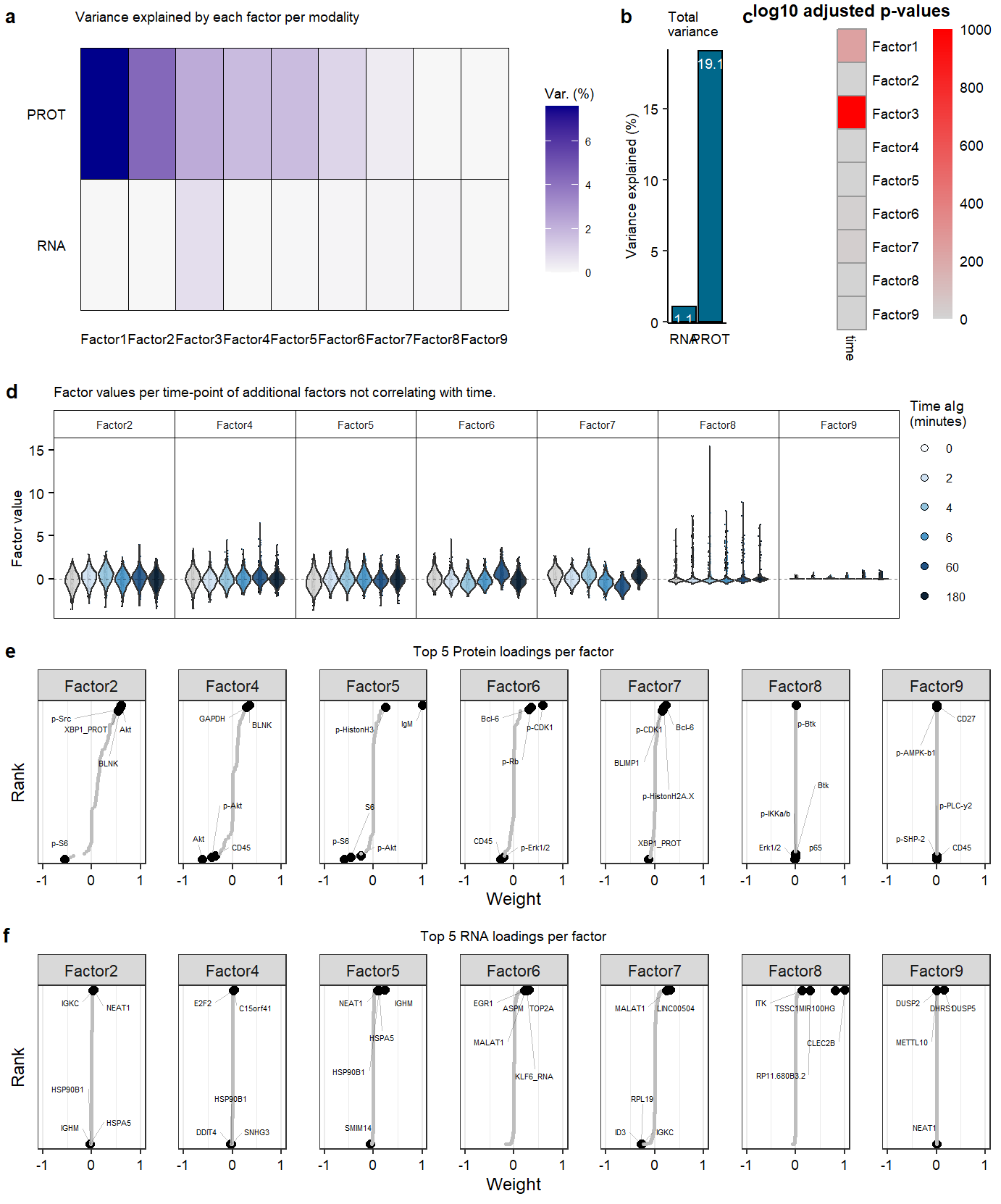 Supplemental Figure. MOFA model additional information
Supplemental Figure. MOFA model additional information
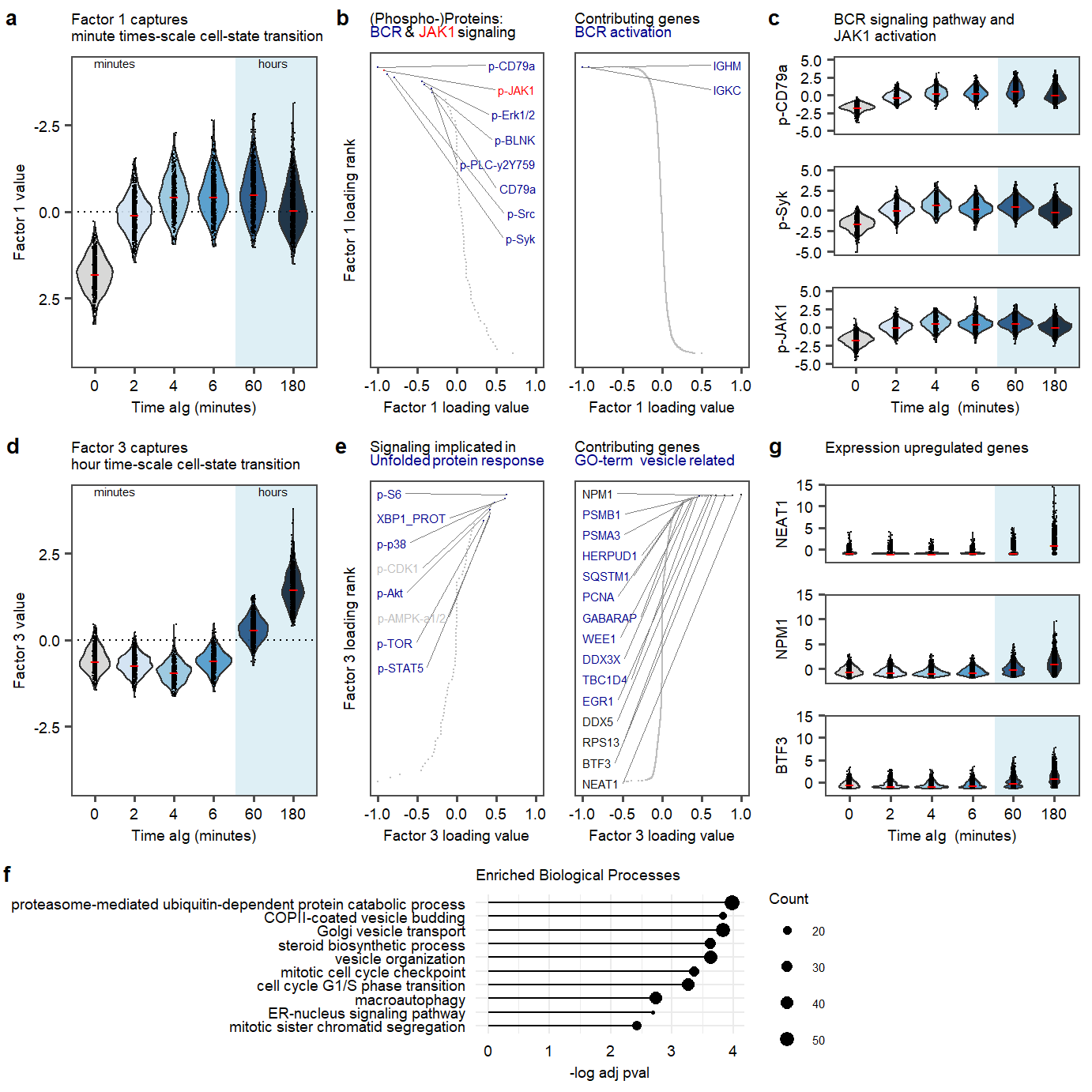 Figure 2. Factor 1 and 3 exploration.
Figure 2. Factor 1 and 3 exploration.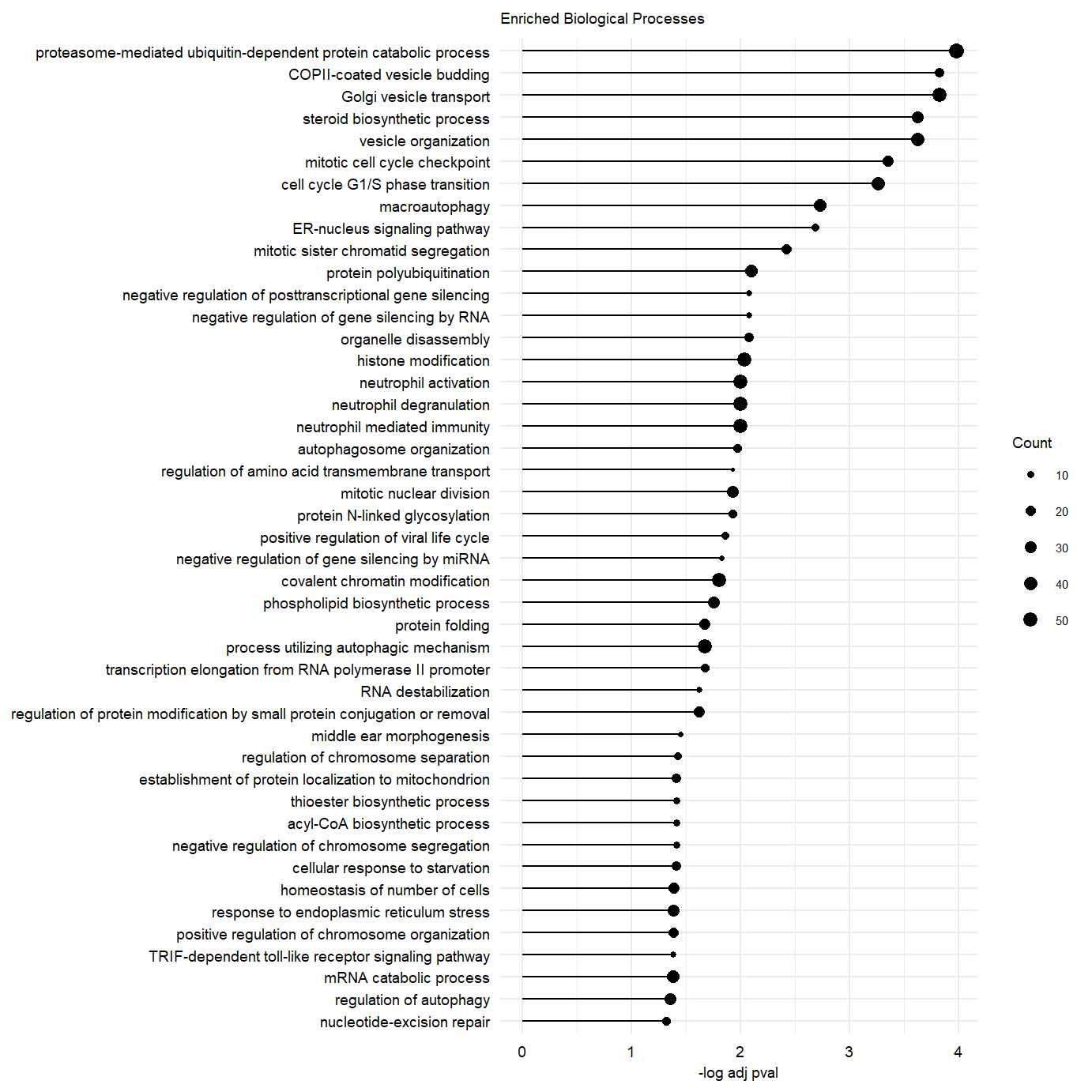 Supplemental Figure. Top 50 enriched gene-sets in postive loadings factor 3.
Supplemental Figure. Top 50 enriched gene-sets in postive loadings factor 3.