Figure 1
UMAP on 7 MOFA factors
## UMAP
plot.umap.data <- plot_dimred(mofa, method="UMAP", color_by = "condition",stroke = 0.001, dot_size =1, alpha = 0.2, return_data = T)
plot.umap.all <- ggplot(plot.umap.data, aes(x=x, y = y, fill = color_by))+
geom_point(size = 0.8, alpha = 0.6, shape = 21, stroke = 0) +
theme_half_open() +
scale_fill_manual(values = colorgradient6_manual, labels = c(labels), name = "Time aIg",)+
theme(legend.position="none")+
scale_x_reverse()+
scale_y_reverse()+
add.textsize +
labs(title = "UMAP on MOFA+ factors shows minute and \nhour time-scale signal transductions", x = "UMAP 1", y = "UMAP 2")
## UMAP legend
legend.umap <- get_legend( ggplot(plot.umap.data, aes(x=x, y = y, fill = color_by))+
geom_point(size = 2, alpha = 1, shape = 21, stroke = 0) +
theme_half_open() +
scale_fill_manual(values = colorgradient6_manual, labels = c(labels), name = "Time aIg \n(minutes)",)+
add.textsize +
labs(title = "UMAP on MOFA+ factors shows minute and \nhour time-scale signal transductions", x = "UMAP 1", "y = UMAP 2"))
legend.umap <- as_ggplot(legend.umap)plot_fig1_umap <- plot_grid(plot.umap.all,legend.umap, labels = c(""), label_size = 10, ncol = 2, rel_widths = c(1, 0.2))
ggsave(plot_fig1_umap, filename = "output/paper_figures/Fig1_UMAP.pdf", width = 68, height = 62, units = "mm", dpi = 300, useDingbats = FALSE)
ggsave(plot_fig1_umap, filename = "output/paper_figures/Fig1_UMAP.png", width = 68, height = 62, units = "mm", dpi = 300)
plot_fig1_umapFigure 1. UMAP of 7 MOFA factors, integrating phospho-protein and RNA measurements
Suppl PCA
seu_combined_selectsamples <- RunPCA(seu_combined_selectsamples,assay = "SCT.RNA", features = genes.variable, verbose = FALSE, ndims.print = 0, reduction.name = "pca.RNA")
seu_combined_selectsamples <- RunPCA(seu_combined_selectsamples, assay = "PROT", features = proteins.all, verbose = FALSE, ndims.print = 0,reduction.name = "pca.PROT")## PCA analysis
plot.PCA.RNA <- DimPlot(seu_combined_selectsamples, reduction = "pca.RNA", group.by = "condition", pt.size =0.2) +
scale_color_manual(values = colorgradient6_manual2, labels = c(labels), name = "Time aIg",)+
labs(title = "RNA PCA separates cells \nat hour time-scale", x= "RNA PC 1", y = " RNA PC 2") +
add.textsize +
theme(legend.position = "none")
plot.PCA.PROT <- DimPlot(seu_combined_selectsamples, reduction = "pca.PROT", group.by = "condition", pt.size = 0.2) +
scale_color_manual(values = colorgradient6_manual2, labels = c(labels), name = "Time aIg",)+
labs(title = "(phospho-)protein PCA separates cells\nat minutes time-scale", x= "Protein PC 1", y = "Protein PC 2") +
add.textsize +
# scale_x_reverse()+
theme(legend.position = "none")
legend <- get_legend(DimPlot(seu_combined_selectsamples, reduction = "pca.RNA", group.by = "condition", pt.size =0.1) +
scale_color_manual(values = colorgradient6_manual2, labels = c(labels), name = "Time aIg",)+
add.textsize)
legend <- as_ggplot(legend)
PCA.PROTPC1.data <- data.frame(rank = 1:80,
protein = names(sort(seu_combined_selectsamples@reductions$pca.PROT@feature.loadings[,1])),
weight.PC1 = sort(seu_combined_selectsamples@reductions$pca.PROT@feature.loadings[,1]),
highlights = c(names(sort(seu_combined_selectsamples@reductions$pca.PROT@feature.loadings[,1]))[1:4],rep("",76))
)
plot.PCA.PROTweightsPC1 <- ggplot(PCA.PROTPC1.data, aes(x=weight.PC1, y = rank, label = highlights)) +
geom_point(size=0.1) +
labs(title = "Top 4 protein loadings \n", x= "Weight Protein PC1") +
geom_point(color = ifelse(PCA.PROTPC1.data$highlights == "", "grey50", "red")) +
geom_text_repel(size = 2, segment.size = 0.25)+
theme_half_open()+
add.textsize +
theme(axis.title.y=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank()
) +
scale_x_reverse()+
scale_y_reverse()
PCA.RNAPC1.data <- data.frame(rank = 1:length(genes.variable),
RNA = names(sort(seu_combined_selectsamples@reductions$pca.RNA@feature.loadings[,1])),
weight.PC1 = sort(seu_combined_selectsamples@reductions$pca.RNA@feature.loadings[,1]),
highlights = c(rep("",(length(genes.variable)-4)),names(sort(seu_combined_selectsamples@reductions$pca.RNA@feature.loadings[,1]))[(length(genes.variable)-3):length(genes.variable)])
)
## Loadings RNA
plot.PCA.RNAweightsPC1 <- ggplot(PCA.RNAPC1.data, aes(x=weight.PC1, y = rank, label = highlights)) +
geom_point(size=0.1) +
labs(title = "Top 4 RNA loadings \n", x= "Weight RNA PC1") +
geom_point(color = ifelse(PCA.RNAPC1.data$highlights == "", "grey50", "red")) +
geom_text_repel(size = 2, segment.size = 0.25)+
theme_half_open()+
add.textsize +
theme(axis.title.y=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)
## ridgeplots
PC1.data <- data.frame(sample = rownames(seu_combined_selectsamples@reductions$pca.PROT@cell.embeddings),
PC1_PROT = seu_combined_selectsamples@reductions$pca.PROT@cell.embeddings[,1],
PC1_RNA = seu_combined_selectsamples@reductions$pca.RNA@cell.embeddings[,1]) %>%
left_join(meta.allcells)
plot_ridge_PC1Prot <- ggplot(PC1.data, aes(x = PC1_PROT, y = condition, fill = condition)) +
scale_fill_manual(values = colorgradient6_manual, labels = c(labels), name = "Time aIg \n(minutes)")+
geom_density_ridges2() +
scale_y_discrete(labels = labels, name = "Time aIg (minutes)")+
scale_x_continuous(name = "PC1 Proteins") +
theme_half_open() +
add.textsize+
theme(legend.position = "none")
plot_ridge_PC1RNA <- ggplot(PC1.data, aes(x = PC1_RNA, y = condition, fill = condition)) +
scale_fill_manual(values = colorgradient6_manual, labels = c(labels), name = "Time aIg \n(minutes)")+
geom_density_ridges2() +
scale_y_discrete(labels = labels, name = "Time aIg (minutes)")+
scale_x_continuous(name = "PC1 RNA") +
theme_half_open() +
add.textsize+
theme(legend.position = "none")Fig1.pca <- plot_grid(plot.PCA.PROT,plot.PCA.RNA,legend, plot_ridge_PC1Prot, plot_ridge_PC1RNA,NULL, plot.PCA.PROTweightsPC1, plot.PCA.RNAweightsPC1, labels = c(panellabels[1:2], "",panellabels[3:4],"",panellabels[5:7]), label_size = 10, ncol = 3, rel_widths = c(0.9,0.9,0.2,0.9,0.9), rel_heights = c(1,0.8,1.3))
ggsave(filename = "output/paper_figures/Suppl_PCA_aIg.pdf", width = 143, height = 170, units = "mm", dpi = 300, useDingbats = FALSE)
ggsave(filename = "output/paper_figures/Suppl_PCA_aIg.png", width = 143, height = 170, units = "mm", dpi = 300)
Fig1.pcaSupplementary Figure. PCA analysis on RNA ór Protein dataset
Suppl MOFA model properties
## Variance per factor
plot.variance.perfactor.all <- plot_variance_explained(mofa, x="factor", y="view") +
add.textsize +
labs(title = "Variance explained by each factor per modality")
## variance total
plot.variance.total <- plot_variance_explained(mofa, x="view", y="factor", plot_total = T)
plot.variance.total <- plot.variance.total[[2]] +
add.textsize +
labs(title = "Total \nvariance") +
geom_text(aes(label=round(R2,1)), vjust=1.6, color="white", size=2.5)
## Significance correlation covariates
plot.heatmap.pval.covariates <- as.ggplot(correlate_factors_with_covariates(mofa,
covariates = c("time"),
factors = 1:mofa@dimensions$K,
fontsize = 7,
cluster_row = F,
cluster_col = F
))+
add.textsize +
theme(axis.text.y=element_blank(),
axis.text.x=element_blank(),
plot.title = element_text(size=7, face = "plain"),
)## Factor values over time
plot.violin.factorall <- plot_factor(mofa,
factor = c(2,4:mofa@dimensions$K),
color_by = "condition",
dot_size = 0.2, # change dot size
dodge = T, # dodge points with different colors
legend = F, # remove legend
add_violin = T, # add violin plots,
violin_alpha = 0.9 # transparency of violin plots
) +
add.textsize +
scale_color_manual(values=c(colorgradient6_manual2, labels = c(labels), name = "Time aIg")) +
scale_fill_manual(values=c(colorgradient6_manual2, labels = c(labels), name = "Time aIg"))+
labs(title = "Factor values per time-point of additional factors not correlating with time." ) +
theme(axis.text.x=element_blank())
## Loadings factors stable protein
plot.rank.PROT.2.4to7 <- plot_weights(mofa,
view = "PROT",
factors = c(1:mofa@dimensions$K),
nfeatures = 3,
text_size = 1.5
) +
add.textsize +
labs(title = "Top 3 Protein loadings per factor") +
theme(axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)
## Loadings factors stable protein
plot.rank.RNA.2.4to7 <- plot_weights(mofa,
view = "RNA",
factors = c(1:mofa@dimensions$K),
nfeatures = 3,
text_size = 1.5
) +
add.textsize +
labs(title = "Top 3 RNA loadings per factor") +
theme(axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)
## correlation time and factors
plot.correlation.covariates <- correlate_factors_with_covariates(mofa,
covariates = c("time"),
factors = mofa@dimensions$K:1,
plot = "r"
)plot.correlation.covariates <- ggcorrplot(plot.correlation.covariates, tl.col = "black", method = "square", lab = TRUE, ggtheme = theme_void, colors = c("#11304C", "white", "#11304C"), lab_size = 2.5) +
add.textsize +
labs(title = "Correlation of \nfactors with \ntime of treatment", y = "") +
scale_y_discrete(labels = "") +
coord_flip() +
theme(axis.text.x=element_text(angle =0,hjust = 0.5),
axis.text.y=element_text(size = 5),
legend.position="none",
plot.title = element_text(hjust = 0.5))Coordinate system already present. Adding new coordinate system, which will replace the existing one.Fig.1.suppl.mofa.row1 <- plot_grid(plot.variance.perfactor.all, plot.variance.total,NULL, plot.heatmap.pval.covariates, labels = c(panellabels[1:3]), label_size = 10, ncol = 4, rel_widths = c(1.35, 0.3,0.25,0.38))
Fig.1.suppl.mofa.row2 <- plot_grid(plot.violin.factorall,legend.umap, labels = panellabels[4], label_size = 10, ncol = 2, rel_widths = c(1,0.1))
Suppl_mofa <- plot_grid(Fig.1.suppl.mofa.row1, Fig.1.suppl.mofa.row2, plot.rank.PROT.2.4to7, plot.rank.RNA.2.4to7, labels = c("","", panellabels[5:6]),label_size = 10, ncol = 1, rel_heights = c(1.45,1,1.1,1.1))
ggsave(Suppl_mofa, filename = "output/paper_figures/Fig2.Suppl_MOFAaIg.pdf", width = 183, height = 220, units = "mm", dpi = 300, useDingbats = FALSE)
ggsave(Suppl_mofa, filename = "output/paper_figures/Fig2.Suppl_MOFAaIg.png", width = 183, height = 220, units = "mm", dpi = 300)
Suppl_mofaSupplementary Figure. MOFA model additional information
Figure 2
Prepare main panels
meta.allcells <- seu_combined_selectsamples@meta.data %>%
mutate(sample = rownames(seu_combined_selectsamples@meta.data))
proteindata <- as.data.frame(t(seu_combined_selectsamples@assays$PROT@scale.data)) %>%
mutate(sample = rownames(t(seu_combined_selectsamples@assays$PROT@scale.data))) %>%
left_join(meta.allcells)
MOFAfactors<- as.data.frame(mofa@expectations$Z) %>%
mutate(sample = rownames(as.data.frame(mofa@expectations$Z)[,1:mofa@dimensions$K]))
MOFAfactors <- left_join(as.data.frame(MOFAfactors), meta.allcells)
weights.prot <- get_weights(mofa, views = "PROT",as.data.frame = TRUE)
topnegprots.factor1 <- weights.prot %>%
filter(factor == "Factor1" & value <= 0) %>%
arrange(value)
topposprots.factor3 <- weights.prot %>%
filter(factor == "Factor3" & value >= 0) %>%
arrange(-value)
weights.RNA <- get_weights(mofa, views = "RNA",as.data.frame = TRUE)## violin plots phospho-proteins highlighted in main
f.violin.fact <- function(data , protein){
ggplot(subset(data) , aes(x = as.factor(condition), y =get(noquote(protein)))) +
annotate("rect",
xmin = 5 - 0.45,
xmax = 6 + 0.6,
ymin = -4.5, ymax = 4.5, fill = "lightblue",
alpha = .4
)+
geom_hline(yintercept=0, linetype='dotted', col = 'black')+
geom_violin(alpha = 0.9,aes(fill = condition))+
geom_jitter(width = 0.05,size = 0.1, fill = "black", shape = 21)+
stat_summary(fun=median, geom="point", shape=95, size=2, inherit.aes = T, position = position_dodge(width = 0.9), color = "red")+
theme_few()+
ylab(paste0(protein)) +
scale_x_discrete(labels = labels, expand = c(0.1,0.1), name = "Time aIg (minutes)") +
scale_y_continuous(expand = c(0,0), name = strsplit(protein, split = "\\.")[[1]][2]) +
scale_color_manual(values = colorgradient6_manual2, labels = c(labels), name = "Time aIg ",)+
scale_fill_manual(values = colorgradient6_manual2, labels = c(labels), name = "Time aIg ",) +
add.textsize +
theme(#axis.title.x=element_blank(),
#axis.text.x=element_blank(),
#axis.ticks.x=element_blank(),
legend.position="none")
}
factors_toplot <- c(colnames(MOFAfactors)[c(1,3)])
for(i in factors_toplot) {
assign(paste0("plot.factor.", i), f.violin.fact(data = MOFAfactors,protein = i))
}
legend.violinfactor <- as_ggplot( get_legend( ggplot(subset(MOFAfactors) , aes(x = as.factor(condition), y =get(noquote(factors_toplot[1])))) +
annotate("rect",
xmin = 5 - 0.45,
xmax = 6 + 0.6,
ymin = -4.5, ymax = 4.5, fill = "lightblue",
alpha = .4
)+
geom_hline(yintercept=0, linetype='dotted', col = 'black')+
geom_violin(alpha = 0.9,aes(fill = condition))+
geom_jitter(width = 0.05,size = 0.1, aes(col = condition), fill = "black", shape = 21)+
stat_summary(fun=median, geom="point", shape=23, size=2, inherit.aes = T, position = position_dodge(width = 0.9), color = "black")+
theme_few()+
ylab(paste0(factors_toplot[1])) +
scale_x_discrete(labels = labels, expand = c(0.1,0.1), name = "Time aIg (minutes)") +
scale_y_continuous(expand = c(0,0), name = strsplit(factors_toplot[1], split = "\\.")[[1]][2]) +
scale_color_manual(values = colorgradient6_manual2, labels = c(labels), name = "Time aIg \n(minutes)",)+
scale_fill_manual(values = colorgradient6_manual2, labels = c(labels), name = "Time aIg \n(minutes)",) +
add.textsize ) )
plot.violin.factor1 <- plot.factor.group1.Factor1 +
scale_y_reverse(expand = c(0,0), name = "Factor 1 value")+
annotate(geom="text", x=1.5, y=-4.3, label="minutes", size = 2,
color="grey3") +
annotate(geom="text", x=5.5, y=-4.3, label="hours", size = 2,
color="grey3") +
labs(title = "Factor 1 captures \nminute times-scale signal transduction")
plot.violin.factor3 <- plot.factor.group1.Factor3 +
annotate(geom="text", x=5.5, y=4.3, label="hours", size = 2,
color="grey3")+
annotate(geom="text", x=1.5, y=4.3, label="minutes", size = 2,
color="grey3") +
scale_y_continuous(expand = c(0,0), name = "Factor 3 value")+
labs(title = "Factor 3 captures \nhour time-scale signal transduction")
#### protein Loadings factor 1
list.bcellpathway.protein <- c("p-CD79a", "p-Syk", "p-Src", "p-Erk1/2", "p-PLC-y2", "p-BLNK","p-PLC-y2Y759","p-PKC-b1", "p-p38", "p-Akt", "p-S6", "p-TOR", "CD79a") # , "p-PLC-y2", "p-PKC-b1", "p-IKKa/b","p-JNK", "p-p38", "p-p65","p-Akt", "p-S6", "p-TOR"
list.top20 <- topnegprots.factor1$feature[1:20]
## protein factor 1
plotdata.rank.PROT.1 <-plot_weights(mofa,
view = "PROT",
factors = c(1),
nfeatures = 15,
text_size = 1,
manual = list(list.top20, list.bcellpathway.protein, "p-JAK1" ),
color_manual = list("grey","blue4","red"),
return_data = TRUE
)
plotdata.rank.PROT.1 <- plotdata.rank.PROT.1 %>%
mutate(Rank = 1:80,
Weight = value,
colorvalue = ifelse(labelling_group == 2 & value <= -0.2,"grey", ifelse(labelling_group == 3 & value <= -0.2, "blue4", ifelse(labelling_group == 4 & value <= -0.2, "red", "grey"))),
highlights = ifelse(labelling_group >= 1 & value <= -0.25, as.character(feature), "")
)
plot.rank.PROT.1 <- ggplot(plotdata.rank.PROT.1, aes(x=Weight, y = Rank, label = highlights)) +
labs(title = "(Phospho-)Proteins: <p><span style='color:blue4'>BCR </span>&<span style='color:red'> JAK1</span> signaling", #<span style='color:blue4'>BCR signaling</span> and <span style='color:red'>p-JAK1</span>
x= "Factor 1 loading value",
y= "Factor 1 loading rank") +
geom_point(size=0.1, color =plotdata.rank.PROT.1$colorvalue, size =1) +
geom_text_repel(size = 2,
segment.size = 0.2,
color =plotdata.rank.PROT.1$colorvalue,
nudge_x = 1 - plotdata.rank.PROT.1$Weight,
direction = "y",
hjust = 1,
segment.color = "grey50")+
theme_few()+
add.textsize +
scale_y_continuous(trans = "reverse") +
add.textsize +
theme(
plot.title = element_markdown()
) +
theme(axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)+
xlim(c(-1,1))
## violin plots phospho-proteins highlighted in main
f.violin.prot <- function(data , protein){
ggplot(subset(data) , aes(x = as.factor(condition), y =get(noquote(protein)))) +
annotate("rect",
xmin = 5 - 0.45,
xmax = 6 + 0.6,
ymin = -5.5, ymax = 5.5, fill = "lightblue",
alpha = .4
)+
geom_violin(alpha = 0.9,aes(fill = condition))+
geom_jitter(width = 0.05,size = 0.1, color = "black")+
stat_summary(fun=median, geom="point", shape=95, size=2, inherit.aes = T, position = position_dodge(width = 0.9), color = "red")+
theme_few()+
ylab(paste0(protein)) +
scale_x_discrete(labels = labels, expand = c(0.1,0.1), name = "Time aIg (minutes)") +
scale_y_continuous(expand = c(0,0), name = protein) +
scale_color_manual(values = colorgradient6_manual2, labels = c(labels), name = "Time aIg ",)+
scale_fill_manual(values = colorgradient6_manual2, labels = c(labels), name = "Time aIg ",) +
add.textsize +
theme(#axis.title.x=element_blank(),
#axis.text.x=element_blank(),
#axis.ticks.x=element_blank(),
legend.position="none")
}
proteins_toplot <- c("p-CD79a","p-Syk","p-JAK1", "IgM")
for(i in proteins_toplot) {
assign(paste0("plot.violin.", i), f.violin.prot(data = proteindata ,protein = i))
}
## Fig 2 bottom row panels
`plot.violin.p-CD79a` <- `plot.violin.p-CD79a` +
labs(title = "BCR signaling pathway and \nJAK1 activation")+
add.textsize+
theme(axis.title.x=element_blank(),
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),)
`plot.violin.p-Syk` <- `plot.violin.p-Syk` +
add.textsize+
theme(axis.title.x=element_blank(),
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),)
`plot.violin.p-JAK1` <-`plot.violin.p-JAK1` +
add.textsize +
labs(y = "<span style='color:red'>p-JAK1</span>") +
theme(axis.title.y = element_markdown()) Enrichment analysis of Factor 3 positive loadings
topposrna.factor3 <- weights.RNA %>%
filter(factor == "Factor3" & value >= 0) %>%
arrange(-value)
rownames(topposrna.factor3) <- topposrna.factor3$feature
### Convert Gene-names to gene IDs (using 'org.Hs.eg.db' library)
topposrna.factor3 <- topposrna.factor3 %>%
mutate(ENTREZID = mapIds(org.Hs.eg.db, as.character(topposrna.factor3$feature), 'ENTREZID', 'SYMBOL'))
listgenes.factor3 <- topposrna.factor3$value
names(listgenes.factor3) <- topposrna.factor3$ENTREZID
go.pb.fct3.pos <- enrichGO(gene = topposrna.factor3$feature,
OrgDb = org.Hs.eg.db,
keyType = 'SYMBOL',
ont = "BP",
pAdjustMethod = "BH")
go.pb.fct3.pos <- simplify(go.pb.fct3.pos, cutoff=0.6, by="p.adjust", select_fun=min)
go.pb.fct3.pos.dplyr <- mutate(go.pb.fct3.pos, richFactor = Count / as.numeric(sub("/\\d+", "", BgRatio)))
## plot main panel
plot.enriched.go.bp.factor3 <- ggplot(go.pb.fct3.pos.dplyr, showCategory = 10,
aes(-log10(p.adjust), fct_reorder(Description, -log10(p.adjust)))) + geom_segment(aes(xend=0, yend = Description)) +
geom_point(aes(size = Count)) +
scale_color_viridis_c(guide=guide_colorbar(reverse=TRUE)) +
scale_size_continuous(range=c(0.5, 3)) +
theme_minimal() +
add.textsize +
xlab("-log adj pval") +
ylab(NULL) +
ggtitle("Enriched Biological Processes") +
theme(legend.position="none")
legend.plot.enriched.go.bp.factor3 <- as.ggplot(get_legend(ggplot(go.pb.fct3.pos.dplyr, showCategory = 10,
aes(-log10(p.adjust), fct_reorder(Description, -log10(p.adjust)))) + geom_segment(aes(xend=0, yend = Description)) +
geom_point(aes(size = Count)) +
scale_color_viridis_c(guide=guide_colorbar(reverse=TRUE)) +
scale_size_continuous(range=c(0.5, 3)) +
theme_minimal() +
add.textsize +
xlab("-log adj pval") +
ylab(NULL) +
ggtitle("Enriched Biological Processes") +
theme(legend.position="right")))
## Plot supplementary
plot.enriched.go.bp.factor3_50 <- ggplot(go.pb.fct3.pos.dplyr, showCategory = 50,
aes(-log10(p.adjust), fct_reorder(Description, -log10(p.adjust)))) + geom_segment(aes(xend=0, yend = Description)) +
geom_point(aes(size = Count)) +
scale_color_viridis_c(guide=guide_colorbar(reverse=TRUE)) +
scale_size_continuous(range=c(0.5, 3)) +
theme_minimal() +
add.textsize +
xlab("-log adj pval") +
ylab(NULL) +
ggtitle("Enriched Biological Processes") message(c("Significance protein folding GO term: \n",go.pb.fct3.pos.dplyr@result[which(go.pb.fct3.pos.dplyr@result$Description == "protein folding"),"p.adjust"]))Significance protein folding GO term:
0.0457794707358827##For RNA loadings panels
topgeneset.fct3<- unlist(str_split(go.pb.fct3.pos.dplyr@result[1:10,"geneID"], pattern = "/"))
topgeneset.fct3 = bitr(topgeneset.fct3, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Hs.eg.db")
## RNA factor 1 loadings
plotdata.rank.RNA.3 <-plot_weights(mofa,
view = "RNA",
factors = c(3),
nfeatures = 5,
text_size = 1,
manual = list(topposrna.factor3$feature[c(1:5)] ,topgeneset.fct3$SYMBOL),
color_manual = list("grey","blue4"),
return_data = TRUE
)
plotdata.rank.RNA.3 <- plotdata.rank.RNA.3 %>%
mutate(Rank = 1:nrow(plotdata.rank.RNA.3),
Weight = value,
colorvalue = ifelse(labelling_group == 3 &value >= 0.25,"blue4", ifelse(labelling_group == 2&value >= 0.25, "grey2", "grey")),
highlights = ifelse(labelling_group >= 1&value >= 0.25, as.character(feature), "")
)
plot.rank.RNA.3 <- ggplot(plotdata.rank.RNA.3, aes(x=Weight, y = Rank, label = highlights)) +
labs(title = "Contributing genes <p><span style='color:blue4'><span style='color:blue4'>GO-term vesicle related </span> ", #<span style='color:blue4'>BCR signaling regulation (find GO term+list todo)</span>
x= "Factor 3 loading value",
y= "Factor 3 loading rank") +
geom_point(size=0.1, color =plotdata.rank.RNA.3$colorvalue) +
geom_text_repel(size = 2,
segment.size = 0.2,
color =plotdata.rank.RNA.3$colorvalue,
nudge_x = -1 - plotdata.rank.RNA.3$Weight,
direction = "y",
hjust = 0,
segment.color = "grey50")+
theme_few()+
add.textsize +
scale_y_continuous() +
add.textsize +
theme(
plot.title = element_markdown()
) +
theme(axis.text.y=element_blank(),
axis.ticks.y=element_blank(),
axis.title.y = element_blank())+
xlim(c(-1,1))
#### Protein loadings factor 3
list.highlight.prot.fct3 <- c("p-p38", "p-Akt", "p-S6", "p-TOR", "XBP1_PROT", "p-STAT5")
list.top20 <- topnegprots.factor1$feature[1:20]
plotdata.rank.PROT.3 <-plot_weights(mofa,
view = "PROT",
factors = c(3),
nfeatures = 30,
text_size = 1,
manual = list(topposprots.factor3$feature[c(1:8)] ,list.highlight.prot.fct3),
color_manual = list("grey","blue4"),
return_data = TRUE
)
plotdata.rank.PROT.3 <- plotdata.rank.PROT.3 %>%
mutate(Rank = 1:80,
Weight = value,
colorvalue = ifelse(labelling_group == 3 &value >= 0.25,"blue4", ifelse(labelling_group == 2&value >= 0.4, "grey", "grey")),
highlights = ifelse(labelling_group >= 1&value >= 0.25, as.character(feature), "")
)
plot.rank.PROT.3 <- ggplot(plotdata.rank.PROT.3, aes(x=Weight, y = Rank, label = highlights)) +
labs(title = "Signaling implicated in <p><span style='color:blue4'>Unfolded protein response</span>",
x= "Factor 3 loading value",
y= "Factor 3 loading rank") +
geom_point(size=0.1, color =plotdata.rank.PROT.3$colorvalue, size =1) +
geom_text_repel(size = 2,
segment.size = 0.2,
color =plotdata.rank.PROT.3$colorvalue,
nudge_x = -1 - plotdata.rank.PROT.3$Weight,
direction = "y",
hjust = 0,
segment.color = "grey50")+
theme_few()+
add.textsize +
scale_y_continuous() +
add.textsize +
theme(
plot.title = element_markdown()
) +
theme(axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)+
xlim(c(-1,1))
#### RNA loadings factor 1
plotdata.rank.RNA.1 <-plot_weights(mofa,
view = "RNA",
factors = c(1),
nfeatures = 2,
text_size = 1,
return_data = TRUE
)
plotdata.rank.RNA.1 <- plotdata.rank.RNA.1 %>%
mutate(Rank = 1:nrow(plotdata.rank.RNA.1),
Weight = value,
colorvalue = ifelse(labelling_group == 1,"blue4", ifelse(labelling_group == 1, "blue4", "grey")),
highlights = ifelse(labelling_group >= 1, as.character(feature), "")
)
plot.rank.RNA.1 <- ggplot(plotdata.rank.RNA.1, aes(x=Weight, y = Rank, label = highlights)) +
labs(title = " Contributing genes<p><span style='color:blue4'>BCR activation</span>",
x= "Factor 1 loading value",
y= "Factor 1 loading rank") +
geom_point(size=0.1, color =plotdata.rank.RNA.1$colorvalue) +
geom_text_repel(size = 2,
segment.size = 0.2,
color =plotdata.rank.RNA.1$colorvalue,
nudge_x = 1 - plotdata.rank.RNA.1$Weight,
direction = "y",
hjust = 1,
segment.color = "grey50") +
theme_few()+
add.textsize +
scale_y_continuous(trans = "reverse") +
add.textsize +
theme(
plot.title = element_markdown()
) +
theme(axis.text.y=element_blank(),
axis.ticks.y=element_blank(),
axis.title.y=element_blank()
)+
xlim(c(-1,1))
#### Violins genes
f.violin.rna <- function(data , protein){
ggplot(subset(data) , aes(x = as.factor(condition), y =get(noquote(protein)))) +
annotate("rect",
xmin = 5 - 0.45,
xmax = 6 + 0.6,
ymin = -3, ymax = 15, fill = "lightblue",
alpha = .4
)+
geom_violin(alpha = 0.9,aes(fill = condition))+
geom_jitter(width = 0.05,size = 0.1, color = "black")+
stat_summary(fun=median, geom="point", shape=95, size=2, inherit.aes = T, position = position_dodge(width = 0.9), color = "red")+
theme_few()+
ylab(paste0(protein)) +
scale_x_discrete(labels = labels, expand = c(0.1,0.1), name = "Time aIg (minutes)") +
scale_y_continuous(expand = c(0,0), name = protein) +
scale_color_manual(values = colorgradient6_manual2, labels = c(labels), name = "Time aIg ",)+
scale_fill_manual(values = colorgradient6_manual2, labels = c(labels), name = "Time aIg ",) +
add.textsize +
theme(#axis.title.x=element_blank(),
#axis.text.x=element_blank(),
#axis.ticks.x=element_blank(),
legend.position="none")
}
rnadata <- as.data.frame(t(seu_combined_selectsamples@assays$SCT.RNA@scale.data)) %>%
mutate(sample = rownames(t(seu_combined_selectsamples@assays$SCT.RNA@scale.data))) %>%
left_join(meta.allcells)
enriched.geneset.posregBcell.fact1 <- c("NPM1", "NEAT1", "BTF3", "IGHM", "IGKC")
##Violin genes
for(i in enriched.geneset.posregBcell.fact1) {
assign(paste0("plot.violin.rna.", i), f.violin.rna(data = rnadata ,protein = i))
}
plot.violin.rna.NEAT1 <- plot.violin.rna.NEAT1 +
labs(title = "Expression upregulated genes\n")+
add.textsize+
theme(axis.title.x=element_blank(),
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),)
plot.violin.rna.NPM1 <- plot.violin.rna.NPM1 +
add.textsize+
theme(axis.title.x=element_blank(),
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),)
Main figure 2
Fig2.row1.violinsprot <- plot_grid(`plot.violin.p-CD79a`, `plot.violin.p-Syk`, `plot.violin.p-JAK1`,labels = c(panellabels[4]), label_size = 10, ncol = 1, rel_heights = c(1.2,0.95,1.2))
Fig2.row1 <- plot_grid(plot.violin.factor1,plot.rank.PROT.1, NULL, Fig2.row1.violinsprot, labels = c(panellabels[1:3], ""), label_size = 10, ncol =4, rel_widths = c(0.8,0.55,0.5,0.8))
Fig2.row2.violinsrna <- plot_grid(plot.violin.rna.NEAT1,plot.violin.rna.NPM1, plot.violin.rna.BTF3,labels = "", label_size = 10, ncol = 1, rel_heights = c(1.2,0.95,1.2))
Fig2.row2 <- plot_grid(plot.violin.factor3,plot.rank.PROT.3,plot.rank.RNA.3,Fig2.row2.violinsrna, labels = c(panellabels[5:6], "", panellabels[8]), label_size = 10, ncol =4, rel_widths = c(0.8,0.55,0.5,0.8))
Fig2.row3 <- plot_grid(plot.enriched.go.bp.factor3,legend.plot.enriched.go.bp.factor3,NULL, labels = panellabels[7], label_size = 10, ncol =3, rel_widths = c(1.8,0.2,0.6))
plot_fig2 <- plot_grid(Fig2.row1, Fig2.row2,Fig2.row3, labels = c("", "", ""), label_size = 10, ncol = 1, rel_heights = c(1,1,0.55))
ggsave(plot_fig2, filename = "output/paper_figures/Fig2.pdf", width = 183, height = 183, units = "mm", dpi = 300, useDingbats = FALSE)
ggsave(plot_fig2, filename = "output/paper_figures/Fig2.png", width = 183, height = 183, units = "mm", dpi = 300)
plot_fig2Figure 2. Factor 1 and 3 exploration.
Suppl Enriched Fact 3
ggsave(plot.enriched.go.bp.factor3_50, filename = "output/paper_figures/Suppl_Enrichm_Fct3.pdf", width = 183, height = 183, units = "mm", dpi = 300, useDingbats = FALSE)
ggsave(plot.enriched.go.bp.factor3_50, filename = "output/paper_figures/Suppl_Enrichm_Fct3.png", width = 183, height = 183, units = "mm", dpi = 300)
plot.enriched.go.bp.factor3_50Supplementary Figure. Top 50 enriched gene-sets in postive loadings factor 3.
sessionInfo()R version 3.6.3 (2020-02-29)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 17763)
Matrix products: default
locale:
[1] LC_COLLATE=English_Netherlands.1252 LC_CTYPE=English_Netherlands.1252
[3] LC_MONETARY=English_Netherlands.1252 LC_NUMERIC=C
[5] LC_TIME=English_Netherlands.1252
attached base packages:
[1] parallel stats4 grid stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] png_0.1-7 forcats_0.5.0
[3] clusterProfiler_3.14.3 clusterProfiler.dplyr_0.0.2
[5] enrichplot_1.6.1 org.Hs.eg.db_3.10.0
[7] AnnotationDbi_1.48.0 IRanges_2.20.2
[9] S4Vectors_0.24.4 Biobase_2.46.0
[11] BiocGenerics_0.32.0 ggridges_0.5.2
[13] cowplot_1.1.0 ggtext_0.1.0
[15] ggplotify_0.0.5 ggcorrplot_0.1.3
[17] ggrepel_0.8.2 ggpubr_0.4.0
[19] scico_1.2.0 MOFA2_1.1
[21] extrafont_0.17 patchwork_1.0.1
[23] RColorBrewer_1.1-2 viridis_0.5.1
[25] viridisLite_0.3.0 ggsci_2.9
[27] sctransform_0.3.1 ggthemes_4.2.0
[29] matrixStats_0.57.0 kableExtra_1.2.1
[31] gridExtra_2.3 Seurat_3.2.2
[33] ggplot2_3.3.2 scales_1.1.1
[35] tidyr_1.1.2 dplyr_1.0.2
[37] stringr_1.4.0 workflowr_1.6.1
loaded via a namespace (and not attached):
[1] reticulate_1.16 tidyselect_1.1.0 RSQLite_2.2.1
[4] htmlwidgets_1.5.2 BiocParallel_1.20.1 Rtsne_0.15
[7] munsell_0.5.0 codetools_0.2-16 ica_1.0-2
[10] future_1.19.1 miniUI_0.1.1.1 withr_2.3.0
[13] GOSemSim_2.12.1 colorspace_1.4-1 knitr_1.30
[16] rstudioapi_0.11 ROCR_1.0-11 ggsignif_0.6.0
[19] tensor_1.5 DOSE_3.12.0 Rttf2pt1_1.3.8
[22] listenv_0.8.0 labeling_0.4.2 git2r_0.27.1
[25] urltools_1.7.3 mnormt_2.0.2 polyclip_1.10-0
[28] farver_2.0.3 bit64_4.0.5 pheatmap_1.0.12
[31] rhdf5_2.30.1 rprojroot_1.3-2 vctrs_0.3.4
[34] generics_0.0.2 xfun_0.18 markdown_1.1
[37] R6_2.4.1 graphlayouts_0.7.0 rsvd_1.0.3
[40] fgsea_1.12.0 spatstat.utils_1.17-0 gridGraphics_0.5-0
[43] DelayedArray_0.12.3 promises_1.1.0 ggraph_2.0.3
[46] gtable_0.3.0 globals_0.13.1 goftest_1.2-2
[49] tidygraph_1.2.0 rlang_0.4.8 splines_3.6.3
[52] rstatix_0.6.0 extrafontdb_1.0 lazyeval_0.2.2
[55] europepmc_0.4 broom_0.7.1 BiocManager_1.30.10
[58] yaml_2.2.1 reshape2_1.4.4 abind_1.4-5
[61] backports_1.1.10 httpuv_1.5.2 qvalue_2.18.0
[64] gridtext_0.1.1 tools_3.6.3 psych_2.0.9
[67] ellipsis_0.3.1 Rcpp_1.0.4.6 plyr_1.8.6
[70] progress_1.2.2 purrr_0.3.4 prettyunits_1.1.1
[73] rpart_4.1-15 deldir_0.1-29 pbapply_1.4-3
[76] zoo_1.8-8 haven_2.3.1 cluster_2.1.0
[79] fs_1.4.1 magrittr_1.5 data.table_1.13.0
[82] DO.db_2.9 openxlsx_4.2.2 triebeard_0.3.0
[85] lmtest_0.9-38 RANN_2.6.1 tmvnsim_1.0-2
[88] whisker_0.4 fitdistrplus_1.1-1 hms_0.5.3
[91] mime_0.9 evaluate_0.14 xtable_1.8-4
[94] rio_0.5.16 readxl_1.3.1 compiler_3.6.3
[97] tibble_3.0.4 KernSmooth_2.23-16 crayon_1.3.4
[100] htmltools_0.5.0 mgcv_1.8-31 later_1.0.0
[103] DBI_1.1.0 tweenr_1.0.1 corrplot_0.84
[106] MASS_7.3-53 rappdirs_0.3.1 Matrix_1.2-18
[109] car_3.0-10 igraph_1.2.6 pkgconfig_2.0.3
[112] rvcheck_0.1.8 foreign_0.8-75 plotly_4.9.2.1
[115] xml2_1.3.2 webshot_0.5.2 rvest_0.3.6
[118] digest_0.6.26 RcppAnnoy_0.0.16 spatstat.data_1.4-3
[121] fastmatch_1.1-0 rmarkdown_2.4 cellranger_1.1.0
[124] leiden_0.3.3 uwot_0.1.8 curl_4.3
[127] shiny_1.5.0 lifecycle_0.2.0 nlme_3.1-144
[130] jsonlite_1.7.1 Rhdf5lib_1.8.0 carData_3.0-4
[133] pillar_1.4.6 lattice_0.20-38 GO.db_3.10.0
[136] fastmap_1.0.1 httr_1.4.2 survival_3.1-8
[139] glue_1.4.2 zip_2.1.1 spatstat_1.64-1
[142] bit_4.0.4 ggforce_0.3.2 stringi_1.4.6
[145] HDF5Array_1.14.4 blob_1.2.1 memoise_1.1.0
[148] irlba_2.3.3 future.apply_1.6.0
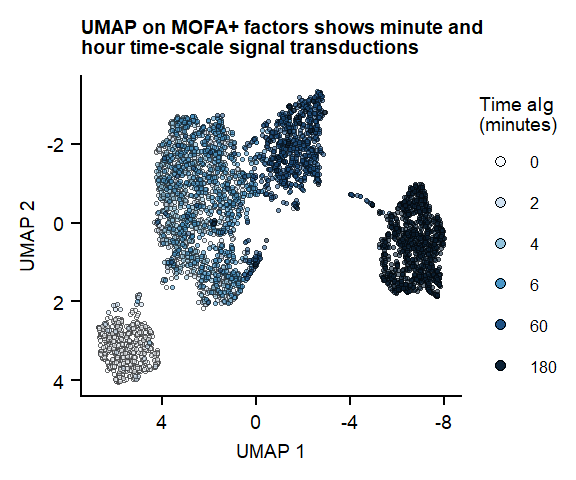 Figure 1. UMAP of 7 MOFA factors, integrating phospho-protein and RNA measurements
Figure 1. UMAP of 7 MOFA factors, integrating phospho-protein and RNA measurements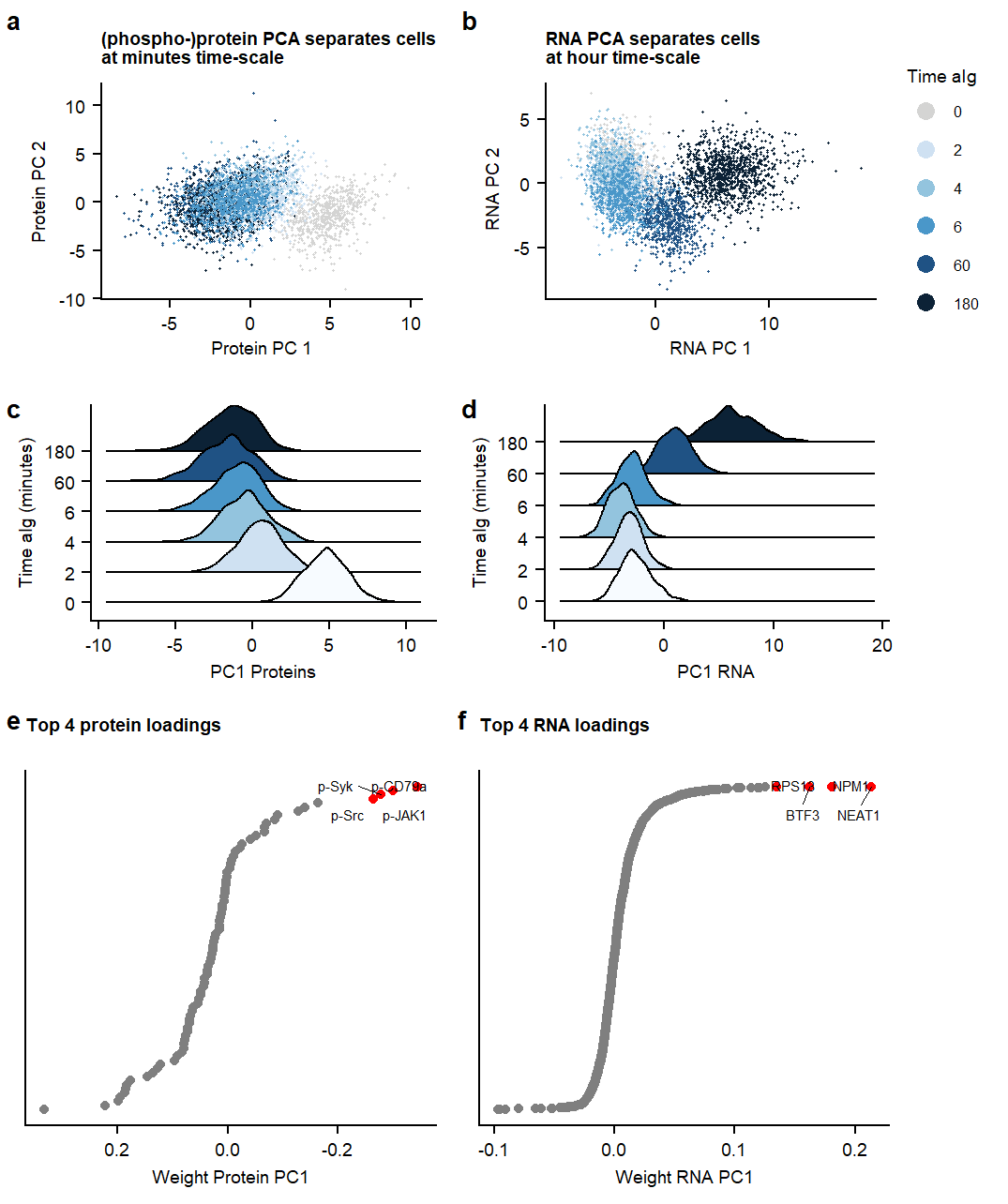 Supplementary Figure. PCA analysis on RNA ór Protein dataset
Supplementary Figure. PCA analysis on RNA ór Protein dataset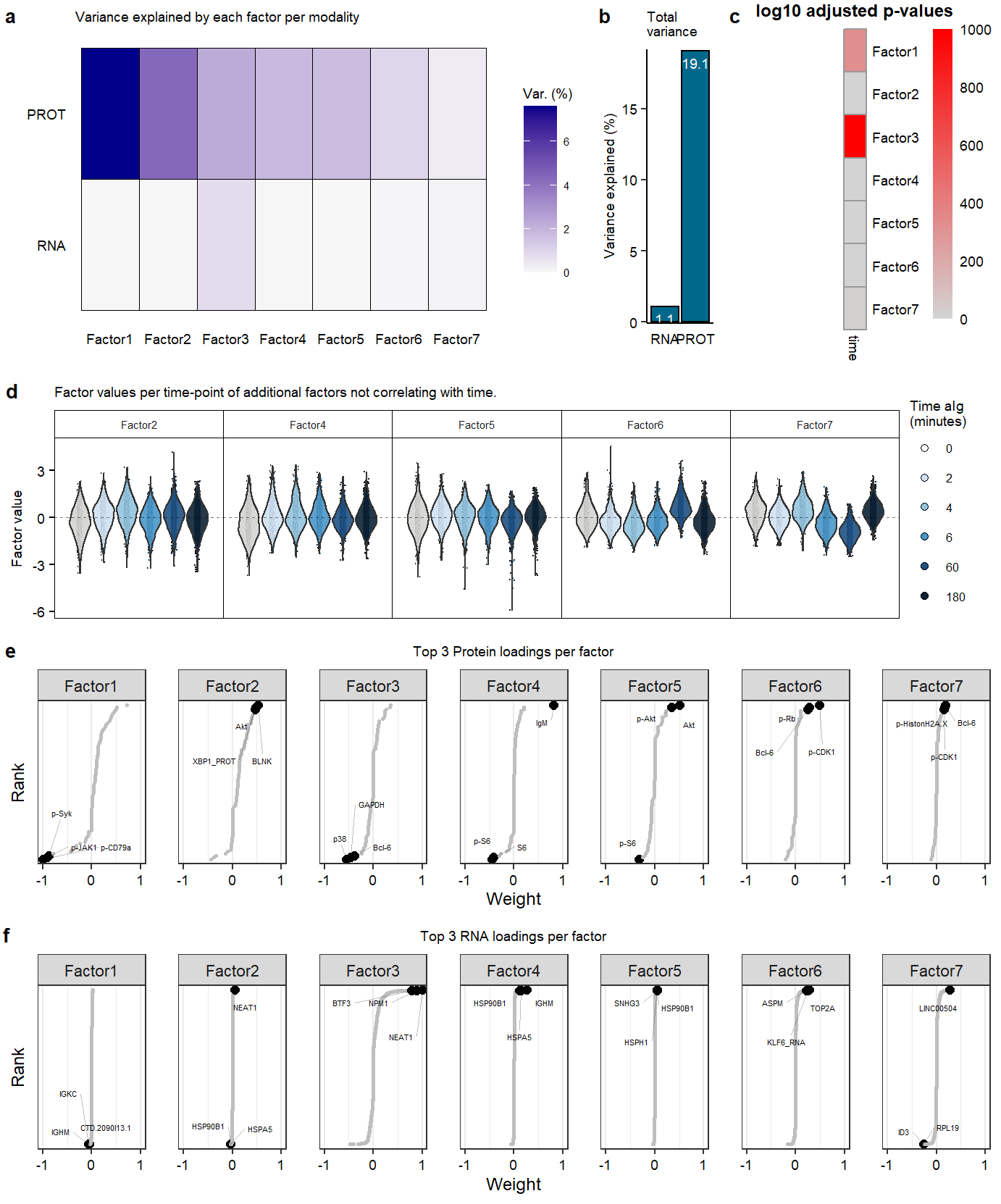 Supplementary Figure. MOFA model additional information
Supplementary Figure. MOFA model additional information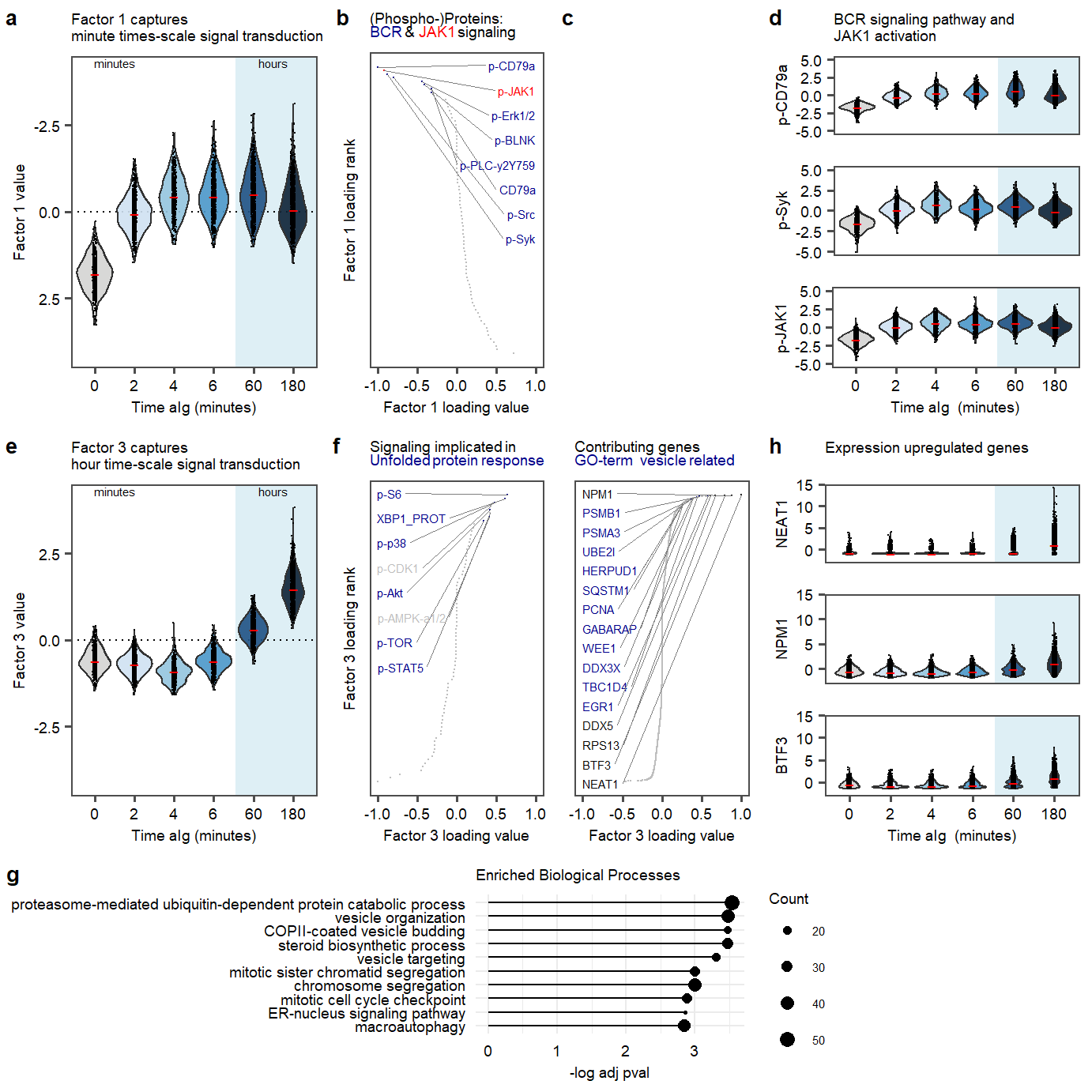 Figure 2. Factor 1 and 3 exploration.
Figure 2. Factor 1 and 3 exploration.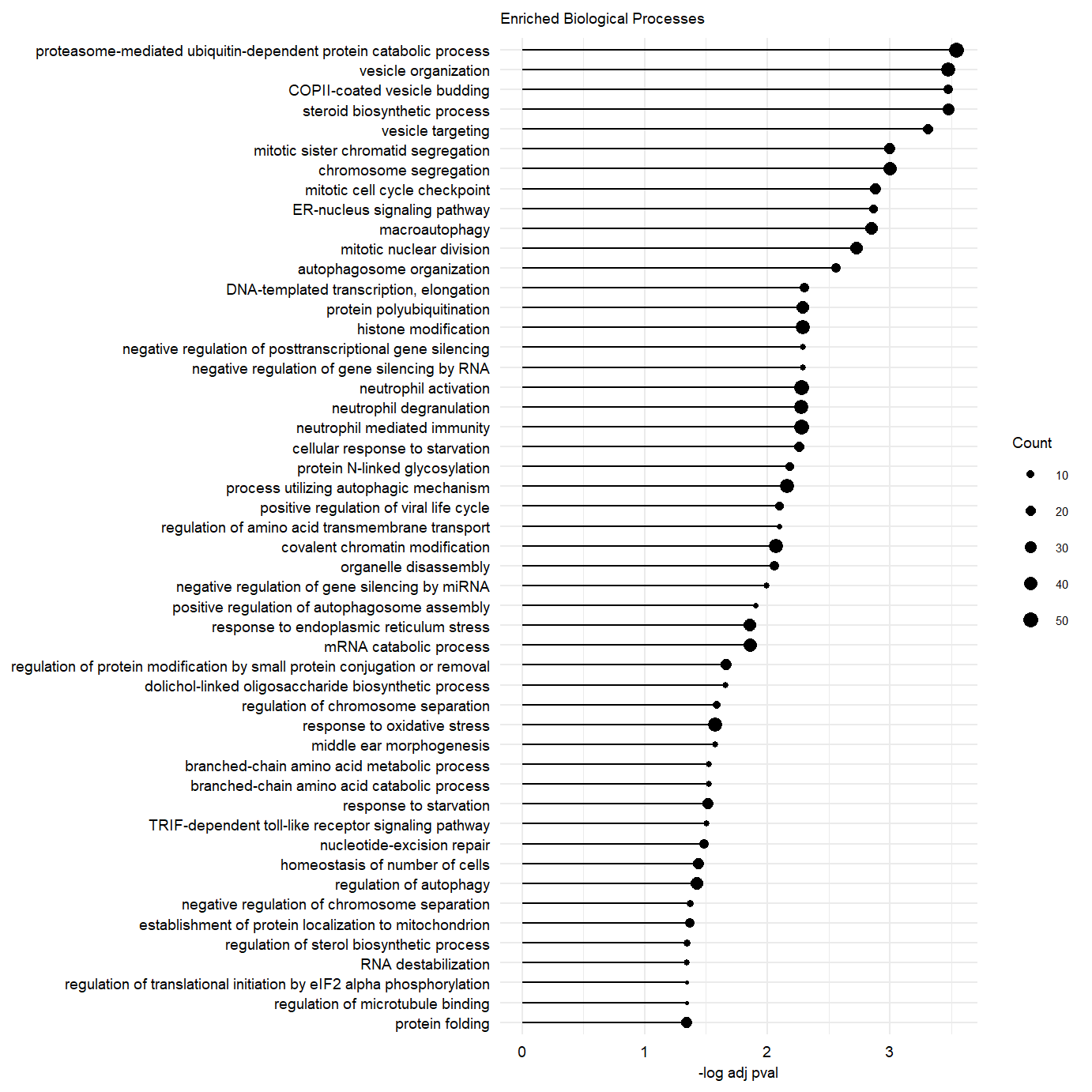 Supplementary Figure. Top 50 enriched gene-sets in postive loadings factor 3.
Supplementary Figure. Top 50 enriched gene-sets in postive loadings factor 3.