lunaphore.heatmaps_pixie.figure3
FloWuenne
2023-08-21
Last updated: 2023-12-06
Checks: 6 1
Knit directory: mi_spatialomics/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230612) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 5dee03d. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis/deprecated/.DS_Store
Ignored: analysis/figure/
Ignored: analysis/molecular_cartography_python/.DS_Store
Ignored: analysis/molecular_cartography_python/figures/
Ignored: analysis/seqIF_python/.DS_Store
Ignored: data/.DS_Store
Ignored: data/140623.calcagno_et_al.seurat_object.rds
Ignored: data/Calcagno2022_int_logNorm_annot.h5Seurat
Ignored: data/mol_cart.heart_regions/
Ignored: data/pixie.cell_table_size_normalized_cell_labels.csv
Ignored: data/results_cts_100.sqm
Ignored: data/seqIF_regions_annotations/
Ignored: data/seurat/
Ignored: figures/.DS_Store
Ignored: figures/Figure_5.pathway_plot.pdf
Ignored: figures/Figure_5.pca_plot.pdf
Ignored: figures/Figure_5.pca_plot.png
Ignored: figures/Figure_5.volcano_plot.pdf
Ignored: figures/Figure_5.vwf_expression_plot.pdf
Ignored: figures/Figure_5.vwf_specificity_plot.pdf
Ignored: figures/Supplementary_figure_3.segmentation_metrics.eps
Ignored: figures/Supplementary_figure_3.segmentation_metrics.png
Ignored: figures/figures.supplementary_figure4.png
Ignored: figures/mol_cart.Figure_2.gains.pdf
Ignored: figures/mol_cart.Figure_2.misty_gains.pdf
Ignored: figures/mol_cart.Myeloid_distribution.png
Ignored: figures/mol_cart.Nppa_distribution.eps
Ignored: figures/mol_cart.Nppa_distribution.pdf
Ignored: figures/mol_cart.Nppa_distribution.png
Ignored: figures/supplementary_figure4.cell_type_distributions.eps
Ignored: figures/supplementary_figure4.cell_type_distributions.png
Ignored: omnipathr-log/
Ignored: output/.DS_Store
Ignored: output/mol_cart.harmony_object.h5Seurat
Ignored: output/mol_cart/
Ignored: output/molkart_cell_types/
Ignored: output/proteomics/
Ignored: output/seqIF/
Ignored: plots/.DS_Store
Ignored: plots/Figure1.umap_plot.pdf
Ignored: plots/Figure3.ccr2_monomacro_regions.pdf
Ignored: plots/Figure3.cell_types_overtimes.pdf
Ignored: plots/Figure3.pixel_clusters_overtimes.pdf
Ignored: plots/mol_cart.Figure_2.ct_percentage.pdf
Ignored: plots/molkart.squidpy.co_occurrence_plot.CMs_Nppa.sample_2d_r1_s1.png
Ignored: plots/molkart.squidpy.co_occurrence_plot.CMs_Nppa.sample_2d_r2_s1.png
Ignored: plots/molkart.squidpy.co_occurrence_plot.CMs_Nppa.sample_control_r1_s1.png
Ignored: plots/molkart.squidpy.co_occurrence_plot.CMs_Nppa.sample_control_r2_s1.png
Ignored: plots/molkart.squidpy.co_occurrence_plot.Endocardial_cells.sample_2d_r1_s1.png
Ignored: plots/molkart.squidpy.co_occurrence_plot.Endocardial_cells.sample_2d_r2_s1.png
Ignored: plots/molkart.squidpy.co_occurrence_plot.Endocardial_cells.sample_control_r1_s1.png
Ignored: plots/molkart.squidpy.co_occurrence_plot.Endocardial_cells.sample_control_r2_s1.png
Ignored: plots/molkart.squidpy.co_occurrence_plot.Myeloid_cells.sample_2d_r1_s1.png
Ignored: plots/molkart.squidpy.co_occurrence_plot.Myeloid_cells.sample_2d_r2_s1.png
Ignored: plots/molkart.squidpy.co_occurrence_plot.Myeloid_cells.sample_control_r1_s1.png
Ignored: plots/molkart.squidpy.co_occurrence_plot.Myeloid_cells.sample_control_r2_s1.png
Ignored: plots/molkart.squidpy.nhood_enrichment_plot.sample_2d_r1_s1.png
Ignored: plots/molkart.squidpy.nhood_enrichment_plot.sample_2d_r2_s1.png
Ignored: plots/molkart.squidpy.nhood_enrichment_plot.sample_control_r1_s1.png
Ignored: plots/molkart.squidpy.nhood_enrichment_plot.sample_control_r2_s1.png
Ignored: references/.DS_Store
Ignored: renv/library/
Ignored: renv/staging/
Untracked files:
Untracked: analysis/deprecated/mol_cart.misty_neighbourhood_analysis.Rmd
Untracked: analysis/deprecated/mol_cart.molkart.process_quantifications_seurat.Rmd
Untracked: analysis/molecular_cartography_python/animation.gif
Untracked: analysis/molecular_cartography_python/kuppe_heart19.h5ad
Untracked: analysis/molecular_cartography_python/mol_cart.plot_points_on_mask.ipynb
Untracked: analysis/molecular_cartography_python/mol_cart.spot_animation.ipynb
Untracked: analysis/molecular_cartography_python/molkart.local_analysis_lianaplus.ipynb
Untracked: analysis/plots/
Untracked: pipeline_configs/
Unstaged changes:
Modified: analysis/data_processing.Rmd
Modified: analysis/figures.Figure1.Rmd
Modified: analysis/figures.Figure2.Rmd
Modified: analysis/figures.deep_visual_proteomics.Rmd
Modified: analysis/figures.supplementary_figure_X.proteomics_qc.Rmd
Modified: analysis/index.Rmd
Deleted: analysis/mol_cart.misty_neighbourhood_analysis.Rmd
Deleted: analysis/mol_cart.molkart.process_quantifications_seurat.Rmd
Modified: analysis/mol_cart.supplementary_figure4.Rmd
Modified: analysis/seqIF.heatmaps_pixie.figure3.Rmd
Modified: analysis/seqIF.region_quantification.Rmd
Modified: analysis/spatialMI_functions.py
Modified: code/functions.R
Modified: figures/Figure_5.eps
Modified: figures/Figure_5.pdf
Modified: figures/Figure_5.png
Modified: figures/Figure_5.svg
Modified: figures/Supplementary_figure_2.png
Modified: figures/Supplementary_figure_5.segmentation_metrics.poster.eps
Modified: figures/Supplementary_figure_X.proteomics.eps
Modified: figures/Supplementary_figure_X.proteomics.png
Modified: plots/Figure1.dotplot.pdf
Modified: plots/molkart.umap_time.png
Modified: references/mh.all.v2023.1.Mm.symbols.sets.rds
Modified: renv.lock
Modified: renv/activate.R
Modified: renv/settings.json
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown
(analysis/seqIF.heatmaps_pixie.figure3.Rmd) and HTML
(docs/seqIF.heatmaps_pixie.figure3.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 5dee03d | FloWuenne | 2023-09-04 | Latest code update. |
| html | 5dee03d | FloWuenne | 2023-09-04 | Latest code update. |
library(pheatmap)
library(data.table)
library(viridis)Loading required package: viridisLitelibrary(RColorBrewer)
library(tidyverse)── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
✔ dplyr 1.1.4 ✔ readr 2.1.4
✔ forcats 1.0.0 ✔ stringr 1.5.1
✔ ggplot2 3.4.4 ✔ tibble 3.2.1
✔ lubridate 1.9.3 ✔ tidyr 1.3.0
✔ purrr 1.0.2 ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::between() masks data.table::between()
✖ dplyr::filter() masks stats::filter()
✖ dplyr::first() masks data.table::first()
✖ lubridate::hour() masks data.table::hour()
✖ lubridate::isoweek() masks data.table::isoweek()
✖ dplyr::lag() masks stats::lag()
✖ dplyr::last() masks data.table::last()
✖ lubridate::mday() masks data.table::mday()
✖ lubridate::minute() masks data.table::minute()
✖ lubridate::month() masks data.table::month()
✖ lubridate::quarter() masks data.table::quarter()
✖ lubridate::second() masks data.table::second()
✖ purrr::transpose() masks data.table::transpose()
✖ lubridate::wday() masks data.table::wday()
✖ lubridate::week() masks data.table::week()
✖ lubridate::yday() masks data.table::yday()
✖ lubridate::year() masks data.table::year()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errorslibrary(pals)
Attaching package: 'pals'
The following objects are masked from 'package:viridis':
cividis, inferno, magma, plasma, turbo, viridis
The following objects are masked from 'package:viridisLite':
cividis, inferno, magma, plasma, turbo, viridislibrary(vroom)
Attaching package: 'vroom'
The following objects are masked from 'package:readr':
as.col_spec, col_character, col_date, col_datetime, col_double,
col_factor, col_guess, col_integer, col_logical, col_number,
col_skip, col_time, cols, cols_condense, cols_only, date_names,
date_names_lang, date_names_langs, default_locale, fwf_cols,
fwf_empty, fwf_positions, fwf_widths, locale, output_column,
problems, speclibrary(plotly)
Attaching package: 'plotly'
The following object is masked from 'package:ggplot2':
last_plot
The following object is masked from 'package:stats':
filter
The following object is masked from 'package:graphics':
layoutpixel_map_color <- c("#0173B2", "#DE8F05", "#029E73", "#D55E00", "#CC78BC",
"#CA9161", "#FBAFE4", "#949494", "#ECE133", "#56B4E9")
cell_cluster_colors <- c("#6b004f","#ff7ed1",
"#018700","#d60000",
"#97ff00","#ffa52f",
"#d55e00","#8c3bff",
"#0000dd","#ff00ff")
names(cell_cluster_colors) <- c("Fibroblasts","Neutrophils",
"Mono / Macros Ccr2+","Smooth muscle cells",
"Macrophages Trem2+","Cardiomyocytes Ankrd1+",
"Endothelial cells","Other Leukocytes",
"Cardiomyocytes","Macrophages Trem2-")
source("./code/functions.R")
Attaching package: 'cowplot'
The following object is masked from 'package:lubridate':
stamp
here() starts at /Users/florian_wuennemann/1_Projects/MI_project/mi_spatialomicsPixel cluster
Pixel cluster maps
avg_pixel_cluster <- fread("/Users/florian_wuennemann/1_Projects/MI_project/Lunaphore/pixie/masked_subset/subset_0.05_wseg/pixel_channel_avg_meta_cluster.csv")
avg_pixel_cluster <- avg_pixel_cluster %>%
subset(pixel_meta_cluster_rename != "background")mat_rownames <- avg_pixel_cluster$pixel_meta_cluster_rename
mat_rownames <- gsub("_","+ ",mat_rownames)
mat_rownames <- paste(mat_rownames,"pixels", sep = " ")
mat_dat <- avg_pixel_cluster %>%
dplyr::select(-c(pixel_meta_cluster,count,pixel_meta_cluster_rename))cap = 3 #hierarchical clustering cap
hclust_coln = "pixel_meta_cluster_rename"
rwb_cols = colorRampPalette(c("royalblue4","white","red4"))(99)
mat_dat = scale(mat_dat)
mat_dat = pmin(mat_dat, cap)
rownames(mat_dat) <- mat_rownames
# Determine breaks
range = max(abs(mat_dat))
breaks = seq(-range, range, length.out=100)
mat_col = data.frame(pixel_cluster = as.factor(mat_rownames))
rownames(mat_col) <- mat_rownames
mat_colors = pixel_map_color[1:length(mat_rownames)]
names(mat_colors) = mat_rownames
mat_colors = list(pixel_cluster = mat_colors)
# Make heatmap
pheatmap(mat_dat,
color = rwb_cols,
border_color = "black",
breaks = breaks,
cluster_rows = TRUE,
cluster_cols = TRUE,
treeheight_col = 25,
treeheight_row = 25,
#treeheight_col = 0,
show_rownames = TRUE,
annotation_row = mat_col,
annotation_colors = mat_colors,
annotation_names_row = FALSE,
annotation_legend = FALSE,
legend = TRUE,
#legend_breaks = c(-3,-2,-1,0,1,2,3),
#legend_labels = c("-3","-2","-1","0","1","2","3"),
main = "",
filename = "./output/seqIF/figure3.pixie_pixel_cluster_heatmap.png",
fontsize = 20,
width = 8,
height = 6)
dev.off()null device
1 Pixel cluster changes over time
## read in pixel cluster counts
pixel_counts = fread("/Users/florian_wuennemann/1_Projects/MI_project/Lunaphore/pixie/masked_subset/subset_0.05_wseg/pixel_counts.all_samples.csv")pixel_cluster_counts_stats <- pixel_counts %>%
subset(Pixel_cluster != "background") %>%
separate("Sample_ID", into = c("time","sample")) %>%
group_by(time,Pixel_cluster) %>%
summarise("n_pixel" = sum(Count)) %>%
mutate("percent" = n_pixel / sum(n_pixel)) %>%
ungroup()`summarise()` has grouped output by 'time'. You can override using the
`.groups` argument.pixel_cluster_counts_stats$time <-factor(pixel_cluster_counts_stats$time,
levels = c("Control","4h","24h","48h"))
pixel_cluster_counts_stats$time_cont <- as.numeric(pixel_cluster_counts_stats$time)#ggplot(cells_over_time, aes(x=time, y=percent, fill=cell_meta_cluster)) +
#geom_bar(stat = "identity", position = "stack",color = "black")
pixel_number_distribution <- ggplot(pixel_cluster_counts_stats,
aes(x=time_cont, y=percent)) +
geom_area(aes(fill = Pixel_cluster), color = "black") +
theme(legend.position = "none",
axis.line = element_blank()) +
scale_fill_manual(values = pixel_map_color) +
scale_x_discrete(expand = c(0,0.1),
name ="Time",
limits=c("Control","4h","24h","48h")) +
labs(y = "% cells")
pixel_number_distribution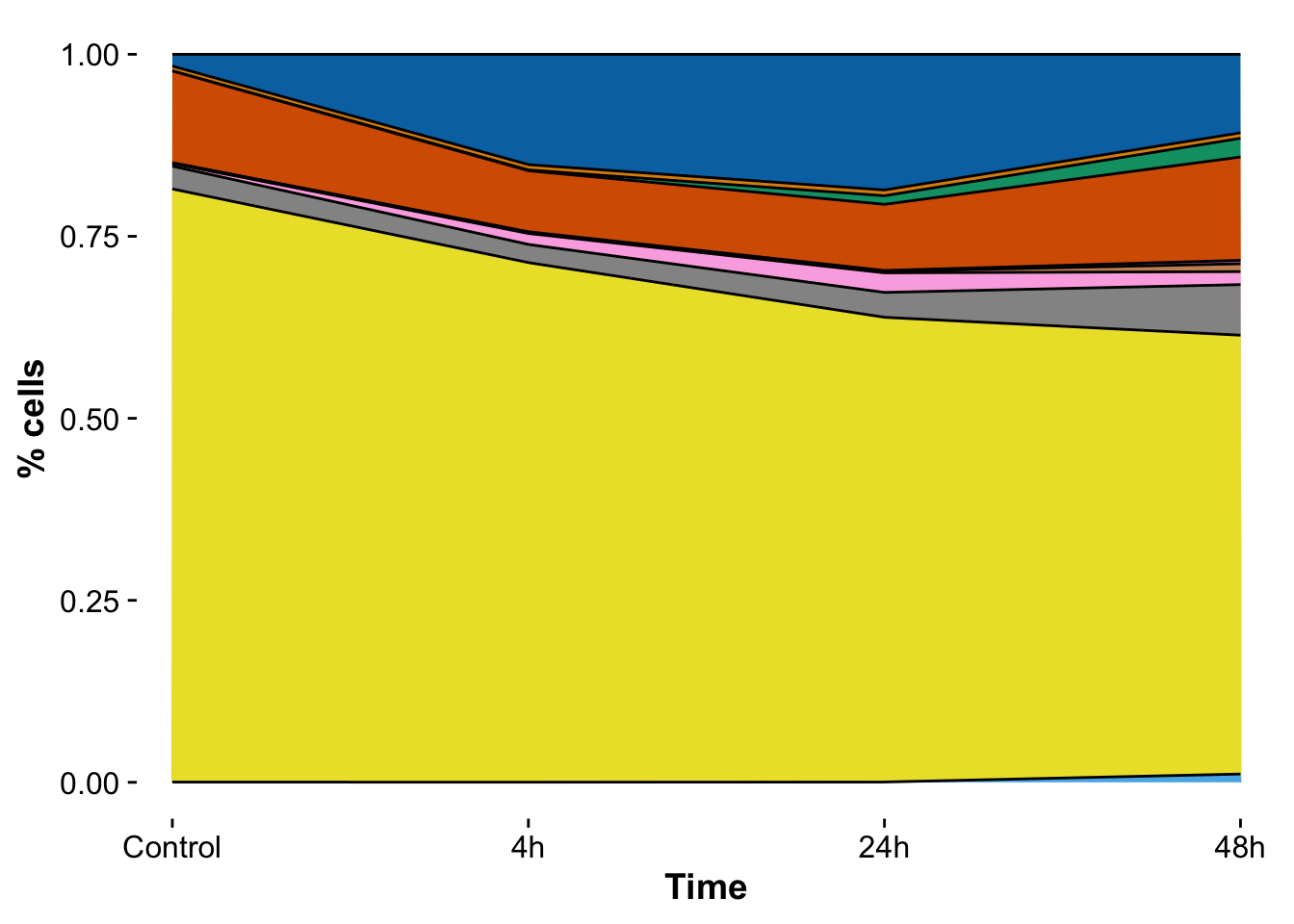
| Version | Author | Date |
|---|---|---|
| 5dee03d | FloWuenne | 2023-09-04 |
save_plot(pixel_number_distribution,
file = "./plots/Figure3.pixel_clusters_overtimes.pdf",
base_height = 3.5,
base_asp = 1)Cell cluster map
Cell cluster tissue maps
avg_cell_cluster <- fread("/Users/florian_wuennemann/1_Projects/MI_project/Lunaphore/pixie/masked_subset/cell_masks_0.05/cell_meta_cluster_count_avg.csv")
colnames(avg_cell_cluster) <- gsub("pixel_meta_cluster_rename_","",colnames(avg_cell_cluster))
avg_cell_cluster <- avg_cell_cluster %>%
subset(cell_meta_cluster_rename != "background") %>%
subset(cell_meta_cluster_rename != "out_of_mask") %>%
dplyr::select(-c(background,count))mat_rownames <- avg_cell_cluster$cell_meta_cluster_rename
mat_rownames <- gsub("_","+ ",mat_rownames)
mat_dat <- avg_cell_cluster %>%
dplyr::select(-c(cell_meta_cluster,cell_meta_cluster_rename))cap = 3 #hierarchical clustering cap
hclust_coln = "pixel_meta_cluster_rename"
rwb_cols = colorRampPalette(c("royalblue4","white","red4"))(99)
mat_dat = scale(mat_dat)
mat_dat = pmin(mat_dat, cap)
rownames(mat_dat) <- mat_rownames
# Determine breaks
range = max(abs(mat_dat))
breaks = seq(-range, range, length.out=100)
## Set color palette
mat_col = data.frame(cell_cluster = as.factor(mat_rownames))
rownames(mat_col) <- mat_rownames
mat_colors = cell_cluster_colors[1:length(mat_rownames)]
names(mat_colors) = mat_rownames
#mat_colors = list(pixel_cluster = mat_colors)
mat_colors = list(cell_cluster = cell_cluster_colors)
# Make heatmap
pheatmap(mat_dat,
color = rwb_cols,
border_color = "black",
breaks = breaks,
cluster_rows = TRUE,
cluster_cols = TRUE,
treeheight_col = 25,
treeheight_row = 25,
#treeheight_col = 0,
show_rownames = TRUE,
annotation_row = mat_col,
annotation_colors = mat_colors,
annotation_names_row = FALSE,
annotation_legend = FALSE,
legend = TRUE,
#legend_breaks = c(-3,-2,-1,0,1,2,3),
#legend_labels = c("-3","-2","-1","0","1","2","3"),
main = "",
filename = "./output/seqIF/figure3.pixie_cell_cluster_heatmap.pdf",
fontsize = 20,
width = 8,
height = 6)Cell cluster changes over time
cells_over_time <- fread("/Users/florian_wuennemann/1_Projects/MI_project/Lunaphore/pixie/masked_subset/cell_masks_0.05/cell_table_size_normalized_cell_labels.csv")
cell_groups_sample <- cells_over_time %>%
separate("fov", into = c("time","sample"), remove = FALSE) %>%
group_by(fov,time,cell_meta_cluster) %>%
tally()
n_cell_plot <- ggplot(cell_groups_sample,aes(cell_meta_cluster,log10(n),color = fov,
label = fov)) +
geom_point(size = 3,aes(shape = time)) +
coord_flip()
ggplotly(n_cell_plot)## Plot distribution of all cell types for a given sample
unique(cells_over_time$cell_meta_cluster) [1] "out_of_mask" "Fibroblasts" "Mono / Macros Ccr2+"
[4] "Cardiomyocytes" "Neutrophils" "background"
[7] "Cardiomyocytes Ankrd1+" "Endothelial cells" "Macrophages Trem2+"
[10] "Smooth muscle cells" "Macrophages Trem2-" "Other Leukocytes" unique(cells_over_time$fov)[1] "24h_86" "24h_83" "Control_13" "Control_12" "4h_97"
[6] "4h_96" "48h_79" "48h_76" "Control_14"cells_over_time_sub <- subset(cells_over_time,fov == "48h_79")
spatial_plot <- ggplot(cells_over_time_sub,aes(X_centroid,Y_centroid,color = cell_meta_cluster)) +
geom_point(size = 0.01) +
facet_wrap(~ cell_meta_cluster) +
labs(title= unique(cells_over_time_sub$fov))
spatial_plot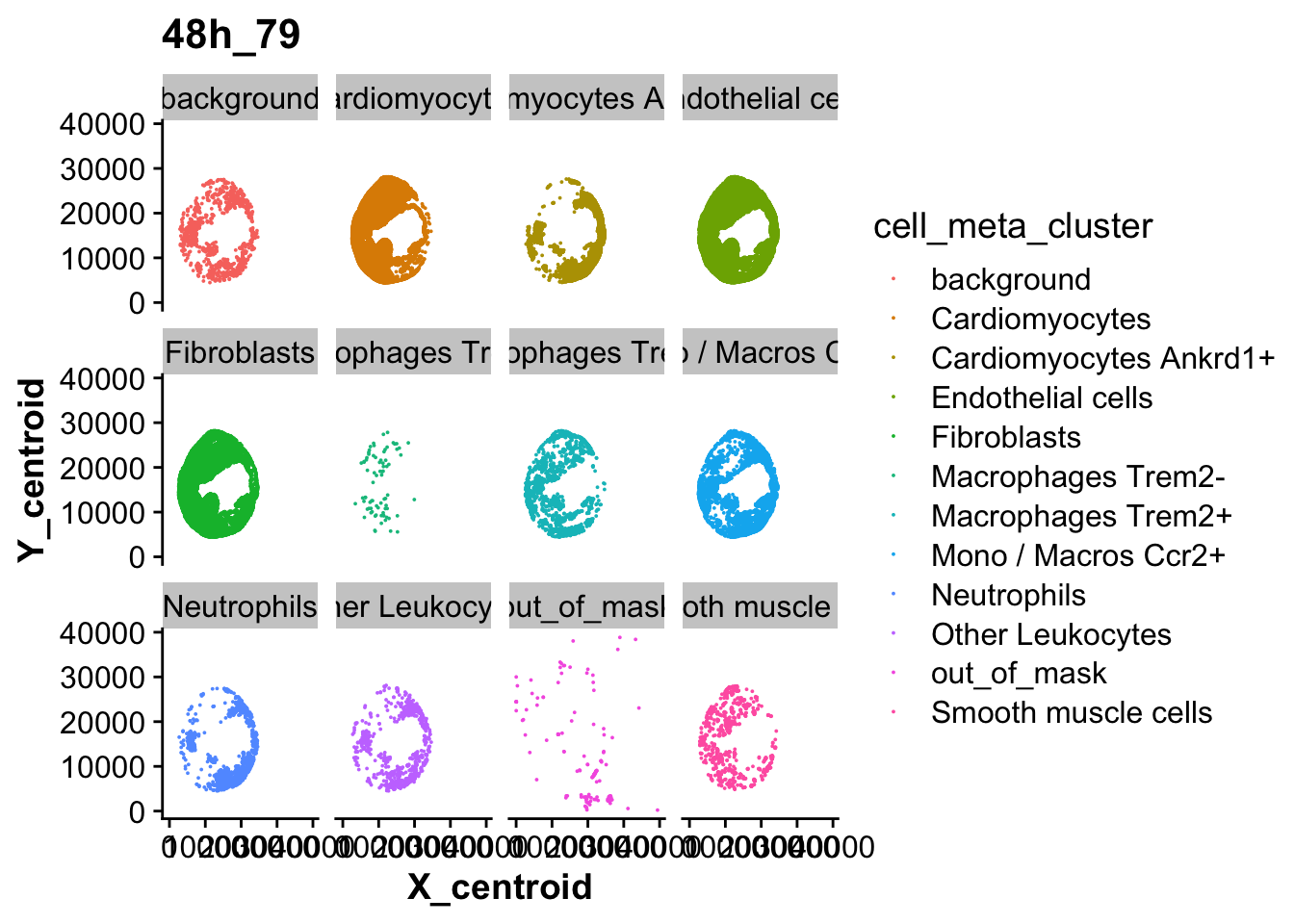
## Plot distribution of 1 cell type over time
# cells_over_time_sub <- subset(cells_over_time,fov %in% c("Control_14","4h_97","24h_86","48h_76"))
cells_over_time_sub <- cells_over_time
pt_size = 0.001
# cells_over_time_sub$fov <- factor(cells_over_time_sub$fov,
# levels = c("Control_14","4h_97","24h_86","48h_76"))
spatial_plot_monos_macros_ccr2 <- ggplot(cells_over_time_sub,aes(X_centroid,Y_centroid)) +
geom_point(data = subset(cells_over_time_sub,cell_meta_cluster == "Cardiomyocytes"),size = pt_size,
color = "darkgrey") +
geom_point(data = subset(cells_over_time_sub,cell_meta_cluster == "Cardiomyocytes Ankrd1+"),size = pt_size,
color = "blue") +
geom_point(data = subset(cells_over_time_sub,cell_meta_cluster == "Neutrophils" | cell_meta_cluster == "Mono / Macros Ccr2+"),size = pt_size,
color = "magenta", alpha = 0.5) +
# geom_point(data = subset(cells_over_time_sub,cell_meta_cluster == "Mono / Macros Ccr2+"),size = pt_size,
# color = "yellow") +
facet_wrap(~ fov)
spatial_plot_monos_macros_ccr2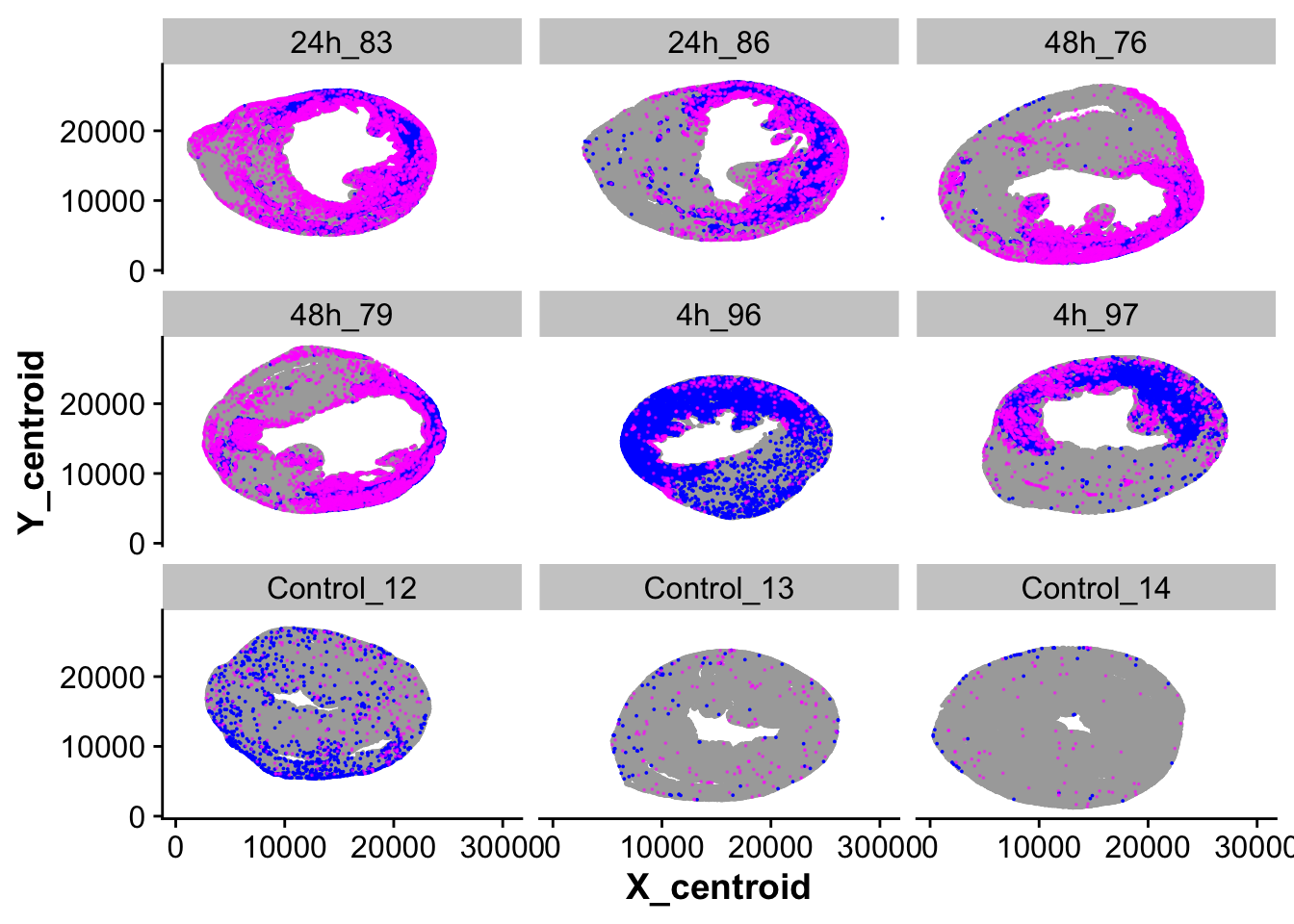
## Color palette
cells_over_time_sub <- cells_over_time %>%
# mutate("cell_type_labels" = if_else(grepl("Macro|Neutro",cell_meta_cluster),"Myeloid cells",cell_meta_cluster))
mutate("cell_type_labels" = cell_meta_cluster)
cells_over_time_sub <- cells_over_time_sub %>%
subset(!cell_type_labels %in% c("background","out_of_mask")) %>%
separate("fov", into = c("time","sample")) %>%
group_by(time,cell_type_labels) %>%
tally() %>%
ungroup()
cells_over_time_sub <- cells_over_time_sub %>%
group_by(time) %>%
mutate("percent" = n / sum(n)) %>%
ungroup()cells_over_time_sub$time <-factor(cells_over_time_sub$time,
levels = c("Control","4h","24h","48h"))
cells_over_time_sub$time_cont <- as.numeric(cells_over_time_sub$time)
cell_number_distribution_bar <- ggplot(cells_over_time_sub, aes(x=time_cont, y=percent, fill= cell_type_labels)) +
#geom_area(aes(fill = cell_type_labels), color = "black") +
geom_bar(stat = "identity", position = "fill", color = "black") +
theme(legend.position = "right",
legend.title = element_blank(),
axis.line = element_blank(),
legend.text=element_text(size=18),
axis.text = element_text(size=18)) +
labs(y = "% cells") +
scale_fill_manual(values = cell_cluster_colors) +
scale_x_discrete(expand = c(0,0.1),
name ="Time",
limits=c("Control","4h","24h","48h")) +
guides(fill=guide_legend(nrow=10,byrow=TRUE))
cell_number_distribution_bar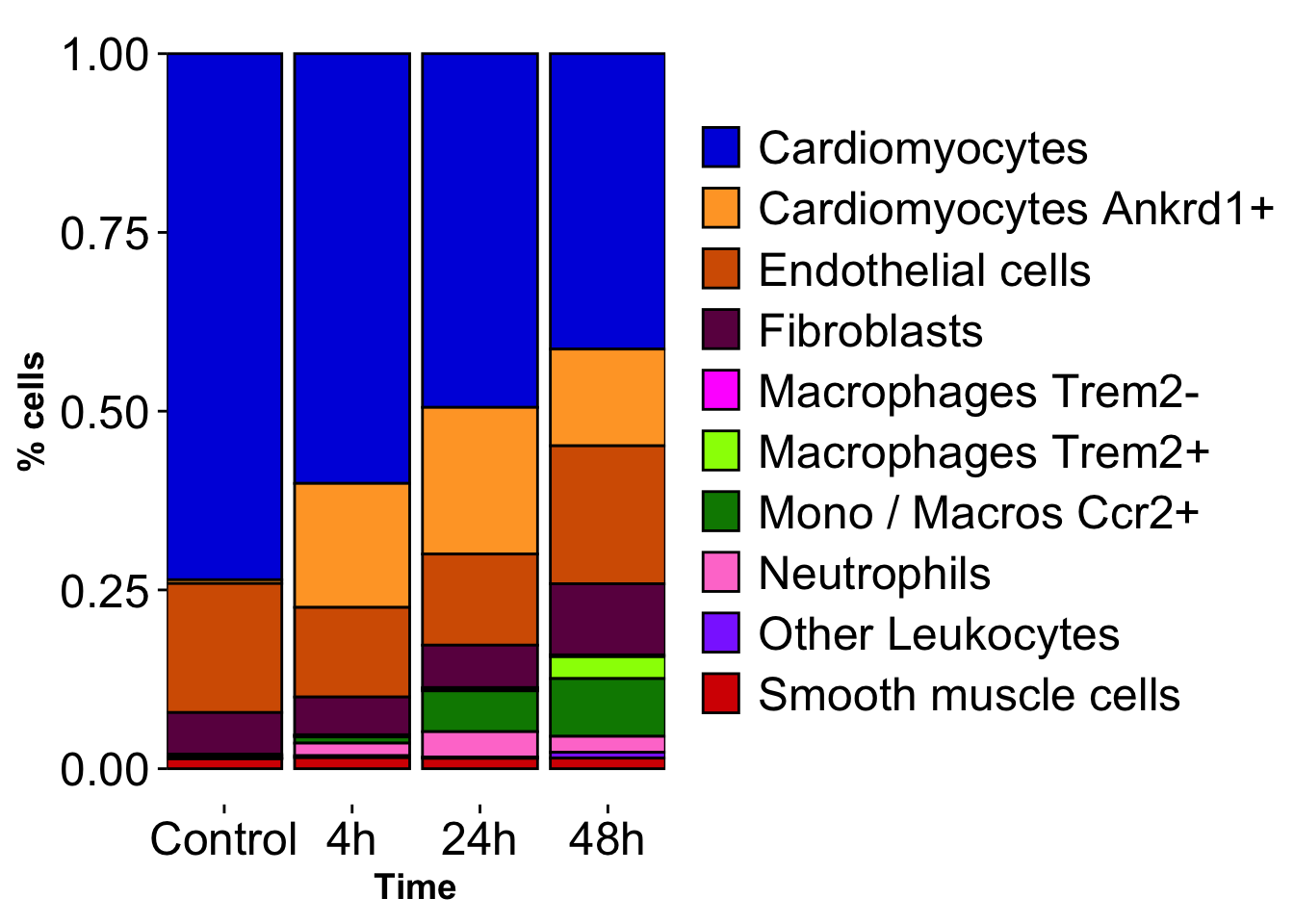
## Continuous plot
# mat_colors = cell_cluster_color[1:length(mat_rownames)]
# cell_number_distribution <- ggplot(cells_over_time_sub, aes(x=time_cont, y=percent)) +
# geom_area(aes(fill = cell_type_labels), color = "black") +
# theme(legend.position = "none",
# axis.line = element_blank()) +
# #scale_fill_manual(values = mat_colors) +
# scale_x_discrete(expand = c(0,0.1),
# name ="Time",
# limits=c("Control","4h","24h","48h")) +
# labs(y = "% cells")
# cell_number_distribution
save_plot(cell_number_distribution_bar,
file = "./plots/Figure3.cell_types_overtimes.pdf",
base_height = 6,
base_asp = 1.3)
sessionInfo()R version 4.3.1 (2023-06-16)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Sonoma 14.1.2
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Europe/Berlin
tzcode source: internal
attached base packages:
[1] stats graphics grDevices datasets utils methods base
other attached packages:
[1] here_1.0.1 ggsci_3.0.0 cowplot_1.1.1 plotly_4.10.3
[5] vroom_1.6.4 pals_1.8 lubridate_1.9.3 forcats_1.0.0
[9] stringr_1.5.1 dplyr_1.1.4 purrr_1.0.2 readr_2.1.4
[13] tidyr_1.3.0 tibble_3.2.1 ggplot2_3.4.4 tidyverse_2.0.0
[17] RColorBrewer_1.1-3 viridis_0.6.4 viridisLite_0.4.2 data.table_1.14.8
[21] pheatmap_1.0.12 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] tidyselect_1.2.0 farver_2.1.1 fastmap_1.1.1
[4] lazyeval_0.2.2 promises_1.2.1 digest_0.6.33
[7] timechange_0.2.0 lifecycle_1.0.4 ellipsis_0.3.2
[10] processx_3.8.2 magrittr_2.0.3 compiler_4.3.1
[13] rlang_1.1.2 sass_0.4.7 tools_4.3.1
[16] utf8_1.2.4 yaml_2.3.7 knitr_1.45
[19] labeling_0.4.3 htmlwidgets_1.6.3 bit_4.0.5
[22] mapproj_1.2.11 withr_2.5.2 grid_4.3.1
[25] fansi_1.0.5 git2r_0.33.0 colorspace_2.1-0
[28] scales_1.3.0 dichromat_2.0-0.1 cli_3.6.1
[31] rmarkdown_2.25 crayon_1.5.2 ragg_1.2.6
[34] generics_0.1.3 rstudioapi_0.15.0 httr_1.4.7
[37] tzdb_0.4.0 cachem_1.0.8 maps_3.4.1.1
[40] BiocManager_1.30.22 vctrs_0.6.5 jsonlite_1.8.8
[43] callr_3.7.3 hms_1.1.3 bit64_4.0.5
[46] crosstalk_1.2.1 systemfonts_1.0.5 jquerylib_0.1.4
[49] glue_1.6.2 ps_1.7.5 stringi_1.8.2
[52] gtable_0.3.4 later_1.3.1 munsell_0.5.0
[55] pillar_1.9.0 htmltools_0.5.7 R6_2.5.1
[58] textshaping_0.3.7 rprojroot_2.0.4 evaluate_0.23
[61] highr_0.10 renv_1.0.3 httpuv_1.6.12
[64] bslib_0.6.1 Rcpp_1.0.11 gridExtra_2.3
[67] whisker_0.4.1 xfun_0.41 fs_1.6.3
[70] getPass_0.2-2 pkgconfig_2.0.3