Sexual selection and sexual size dimorphism: a meta-analysis of comparative studies
Main analyses
Lennart Winkler1, Robert P
Freckleton2, Tamas Szekely3, 4 &
Tim Janicke1,5
1Applied Zoology,
Technical University Dresden 2Department of
Zoology, University of Oxford, South Parks Road, Oxford OX1 3PS,
UK
3Milner Centre for Evolution, University of Bath,
Bath, UK84Department of Evolutionary Zoology and
Human Behaviour, University of Debrecen, Debrecen, Hungary
5Centre d’Écologie Fonctionnelle et Évolutive, UMR 5175,
CNRS, Université de Montpellier
Last updated: 2023-05-24
Checks: 7 0
Knit directory:
SSD_and_sexual_selection_2023/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230430) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version c1d3311. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: analysis/figure/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/index.Rmd) and HTML
(docs/index.html) files. If you’ve configured a remote Git
repository (see ?wflow_git_remote), click on the hyperlinks
in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | c1d3311 | LennartWinkler | 2023-05-24 | wflow_publish(all = T) |
| html | 71d667c | LennartWinkler | 2023-05-03 | Build site. |
| Rmd | 378a58c | LennartWinkler | 2023-05-03 | wflow_publish(republish = TRUE, all = T) |
| html | 085f45f | LennartWinkler | 2023-05-03 | Build site. |
| Rmd | 85c143e | LennartWinkler | 2023-05-03 | update |
| html | 8edd942 | LennartWinkler | 2023-05-01 | Build site. |
| Rmd | d131cd9 | LennartWinkler | 2023-05-01 | wflow_publish(republish = TRUE, all = T) |
| Rmd | 57ca562 | LennartWinkler | 2023-04-30 | update |
| html | 054a12b | LennartWinkler | 2023-04-30 | Build site. |
| Rmd | 1a5561c | LennartWinkler | 2023-04-30 | wflow_publish(all = T) |
| Rmd | 4482c88 | LennartWinkler | 2023-04-30 | Start workflowr project. |
Supplementary material reporting R code for the manuscript ‘Sexual selection and sexual size dimorphism: a meta-analysis of comparative studies’. Additional analyses excluding studies that did not control for phylogenetic non-independence (Supplement 2) can be found at: https://https://lennartwinkler.github.io/SSD_and_sexual_selection_2023/index2.html # Load and prepare data Before we started the analyses, we loaded all necessary packages and data.
rm(list = ls()) # Clear work environment
# Load R-packages ####
list_of_packages=cbind('ape','matrixcalc','metafor','Matrix','MASS','pwr','psych','multcomp','data.table','ggplot2','RColorBrewer','MCMCglmm','ggdist','cowplot','PupillometryR','dplyr','wesanderson')
lapply(list_of_packages, require, character.only = TRUE)
# Load data set ####
MetaData <- read.csv("./data/Supplement4_SexSelSSD_V01.csv", sep=";", header=TRUE) # Load data set
N_Studies <- length(summary(as.factor(MetaData$Study_ID))) # Number of included primary studies
Tree<- read.tree("./data/Supplement6_SexSelSSD_V01.txt") # Load phylogenetic tree
# Prune phylogenetic tree
MetaData_Class_Data <- unique(MetaData$Class)
Tree_Class<-drop.tip(Tree, Tree$tip.label[-na.omit(match(MetaData_Class_Data, Tree$tip.label))])
forcedC_Moderators <- as.matrix(forceSymmetric(vcv(Tree_Class, corr=TRUE)))
# Order moderator levels
MetaData$SexSel_Mode=as.factor(MetaData$SexSel_Mode)
MetaData$SexSel_Mode=relevel(MetaData$SexSel_Mode,c("post-copulatory"))
MetaData$SexSel_Mode=relevel(MetaData$SexSel_Mode,c("pre-copulatory"))
MetaData$SexSel_Sex=as.factor(MetaData$SexSel_Sex)
MetaData$SexSel_Sex=relevel(MetaData$SexSel_Sex,c("Male"))
# Set figure theme and colors
theme=theme(panel.border = element_blank(),
panel.background = element_blank(),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
legend.position = c(0.2,0.5),
legend.title = element_blank(),
legend.text = element_text(colour="black", size=12),
axis.line.x = element_line(colour = "black", size = 1),
axis.line.y = element_line(colour = "black", size = 1),
axis.text.x = element_text(face="plain", color="black", size=16, angle=0),
axis.text.y = element_text(face="plain", color="black", size=16, angle=0),
axis.title.x = element_text(size=16,face="plain", margin = margin(r=0,10,0,0)),
axis.title.y = element_text(size=16,face="plain", margin = margin(r=10,0,0,0)),
axis.ticks = element_line(size = 1),
axis.ticks.length = unit(.3, "cm"))
colpal=c("#4DAF4A","#377EB8","#E41A1C")
colpal2=brewer.pal(7, 'Dark2')
colpal3=wes_palette('FantasticFox1', 9, type = c("continuous"))
colpal4=c("grey50","grey65")
Meta_col=c('grey85','grey50','grey20','black')
# Global models ####
# Phylogenetic Model
Model_REML_Null = rma.mv(r ~ 1, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_Null)
# Non-phylogenetic Model
Model_cREML_Null = rma.mv(r ~ 1, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_Null)Global model Version 1
First, we tested if increasing sexual selection (estimated via diverse proxies; see Table S1) correlated with the degree of SSD. To this end we ran a global model on the absolute correlation coefficients extracted from primary studies (i.e. increasing sexual selection could correlate with a female- or male-bias in SSD, with all resulting correlation coefficients being positive). First, we ran a global model including the phylogeny:
# Set all correlation coeficients as positive
MetaData$absSSDcor=NA
MetaData$absSSDcor=abs(MetaData$r)
# Phylogenetic Model
Model_REML_Null = rma.mv(absSSDcor ~ 1, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_Null)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
8.8296 -17.6591 -9.6591 0.0642 -9.1528
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0193 0.1388 51 no Study_ID no
sigma^2.2 0.0146 0.1207 85 no Index no
sigma^2.3 0.0100 0.1002 9 no Class yes
Test for Heterogeneity:
Q(df = 84) = 722.9083, p-val < .0001
Model Results:
estimate se zval pval ci.lb ci.ub
0.3529 0.0674 5.2396 <.0001 0.2209 0.4849 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Second, we ran a global model without the phylogeny:
# Non-phylogenetic Model
Model_cREML_Null = rma.mv(absSSDcor ~ 1, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_Null)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
7.8506 -15.7013 -9.7013 -2.4088 -9.4013
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0225 0.1501 51 no Study_ID
sigma^2.2 0.0159 0.1261 85 no Index
Test for Heterogeneity:
Q(df = 84) = 722.9083, p-val < .0001
Model Results:
estimate se zval pval ci.lb ci.ub
0.3833 0.0293 13.1019 <.0001 0.3260 0.4406 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Global models Version 2
Next, we addressed the question if increasing sexual selection correlated with an increasingly male-biased SSD. For this we ran a global model including an observation-level index and the study identifier as random termson correlation coefficients that were positive if increasing sexual selection correlated with an increasingly male-biased SSD, but negative if increasing sexual selection correlated with an increasingly female-biased SSD. First, we ran a global model including the phylogeny:
Model_REML_Null = rma.mv(r ~ 1, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_Null)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
-23.9950 47.9900 55.9900 65.7133 56.4964
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0252 0.1586 51 no Study_ID no
sigma^2.2 0.0617 0.2485 85 no Index no
sigma^2.3 0.0161 0.1269 9 no Class yes
Test for Heterogeneity:
Q(df = 84) = 1447.8890, p-val < .0001
Model Results:
estimate se zval pval ci.lb ci.ub
0.2898 0.0875 3.3109 0.0009 0.1182 0.4613 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Second, we ran a global model without the phylogeny:
Model_cREML_Null = rma.mv(r ~ 1, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_Null)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
-24.9810 49.9619 55.9619 63.2544 56.2619
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0336 0.1834 51 no Study_ID
sigma^2.2 0.0622 0.2494 85 no Index
Test for Heterogeneity:
Q(df = 84) = 1447.8890, p-val < .0001
Model Results:
estimate se zval pval ci.lb ci.ub
0.2823 0.0410 6.8804 <.0001 0.2019 0.3627 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Moderator tests for phylogenetic models
Next, we ran a series of models that test the effect of different moderators. Again we started with models including the phylogeny.
Sexual selection mode
The first model explores the effect of the sexual selection mode (i.e. pre-copulatory, post-copulatory or both):
MetaData$SexSel_Mode=relevel(MetaData$SexSel_Mode,c("pre-copulatory"))
Model_REML_by_SexSelMode = rma.mv(r ~ factor(SexSel_Mode), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelMode)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
-11.4578 22.9157 34.9157 49.3560 36.0357
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0169 0.1300 51 no Study_ID no
sigma^2.2 0.0438 0.2093 85 no Index no
sigma^2.3 0.0164 0.1281 9 no Class yes
Test for Residual Heterogeneity:
QE(df = 82) = 1230.9203, p-val < .0001
Test of Moderators (coefficients 2:3):
QM(df = 2) = 30.1226, p-val < .0001
Model Results:
estimate se zval pval ci.lb
intrcpt 0.2671 0.0897 2.9790 0.0029 0.0914
factor(SexSel_Mode)post-copulatory -0.4664 0.1109 -4.2062 <.0001 -0.6837
factor(SexSel_Mode)both 0.1505 0.0654 2.3026 0.0213 0.0224
ci.ub
intrcpt 0.4429 **
factor(SexSel_Mode)post-copulatory -0.2491 ***
factor(SexSel_Mode)both 0.2786 *
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$SexSel_Mode=relevel(MetaData$SexSel_Mode,c("post-copulatory"))
Model_REML_by_SexSelMode2 = rma.mv(r ~ factor(SexSel_Mode), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelMode2)
MetaData$SexSel_Mode=relevel(MetaData$SexSel_Mode,c("both"))
Model_REML_by_SexSelMode3 = rma.mv(r ~ factor(SexSel_Mode), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelMode3)Finally, we computed FDR corrected p-values:
tab1=as.data.frame(round(p.adjust(c(0.0029, 0.1203, .0001), method = 'fdr'),digit=3),row.names=cbind("Pre-copulatory","Post-copulatory","Both"))
colnames(tab1)<-cbind('P-value')
tab1 P-value
Pre-copulatory 0.004
Post-copulatory 0.120
Both 0.000Plot sexual selection mode (Figure 2)
Here we plot the sexual selection mode moderator:
MetaData$SexSel_Mode=factor(MetaData$SexSel_Mode, levels = c("both","post-copulatory" ,"pre-copulatory"))
ggplot(MetaData, aes(x=SexSel_Mode, y=r, fill = SexSel_Mode, colour = SexSel_Mode)) +
geom_hline(yintercept=0, linetype="longdash", color = "black", linewidth=1)+
geom_flat_violin(position = position_nudge(x = 0.25, y = 0),adjust =1, trim = F,alpha=0.6)+
geom_point(position = position_jitter(width = .1), size = 2.5,alpha=0.6,stroke=0,shape=19)+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelMode$b[1,1], x=3.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelMode2$b[1,1], x=2.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelMode3$b[1,1], x=1.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelMode$ci.lb[1], x=3.25, xend= 3.25, yend= Model_REML_by_SexSelMode$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelMode2$ci.lb[1], x=2.25, xend= 2.25, yend= Model_REML_by_SexSelMode2$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelMode3$ci.lb[1], x=1.25, xend= 1.25, yend= Model_REML_by_SexSelMode3$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
ylab(expression(paste("Effect size (", italic("r"),')')))+xlab('Sexual selection mode')+coord_flip()+guides(fill = FALSE, colour = FALSE) +
scale_color_manual(values =colpal)+
scale_fill_manual(values =colpal)+
scale_x_discrete(labels=c("Both","Post-copulatory" ,"Pre-copulatory"),expand=c(.1,0))+
annotate("text", x=1, y=1.2, label= "n = 37",size=4.5) +
annotate("text", x=2, y=1.2, label= "n = 9",size=4.5) +
annotate("text", x=3, y=1.2, label= "n = 39",size=4.5) + theme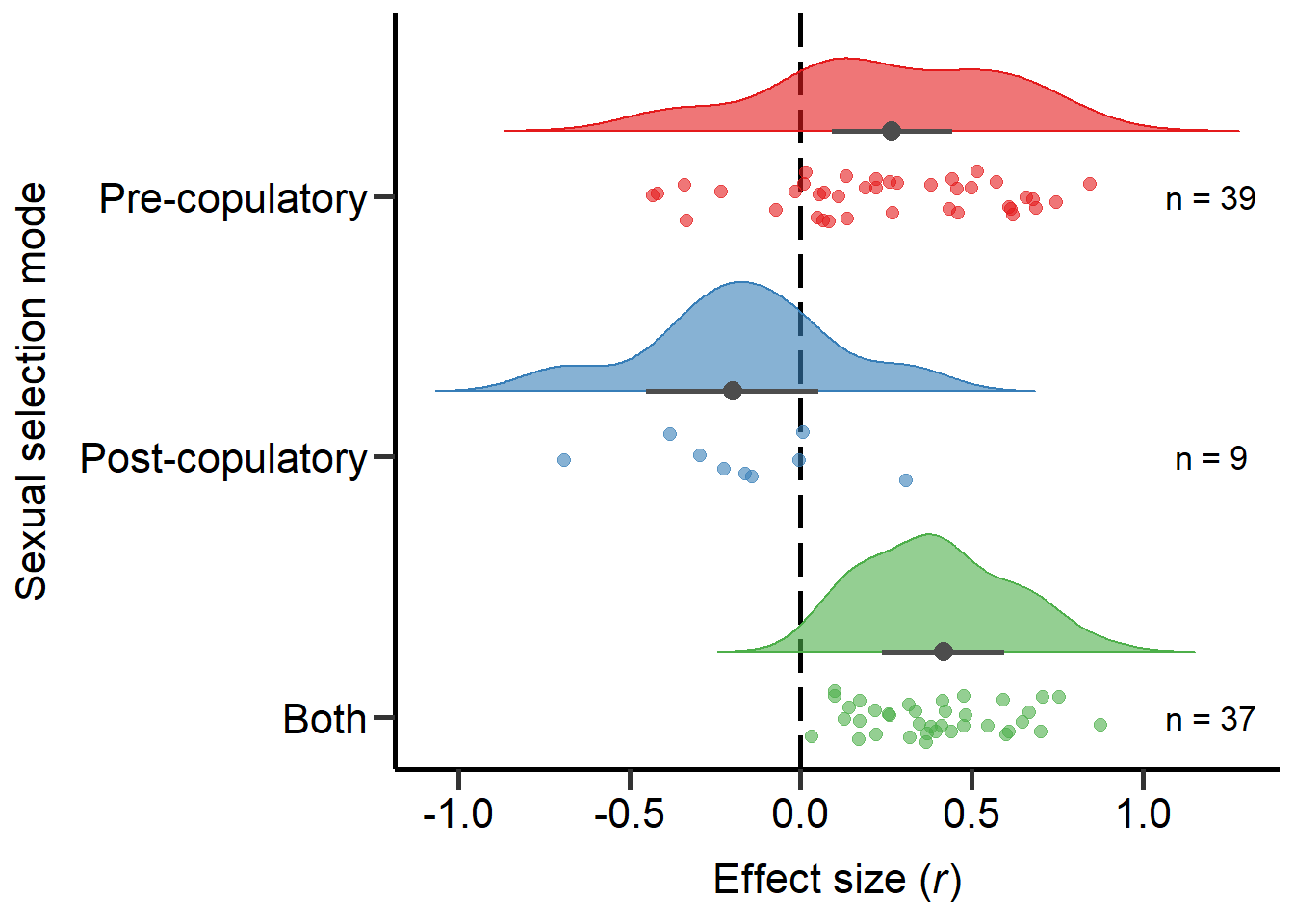
Figure 2: Raincloud plot of correlation coefficients between SSD and the modes of sexual selection proxies (i.e. pre-copulatory, post-copulatory or both) including sample sizes and estimates with 95%CI from phylogenetic model.
Sexual selection category
Next we explored the effect of the sexual selection category (i.e. density, mating system, operational sex ratio (OSR), post-mating competition, pre-mating competition, trait-based, other):
MetaData$SexSel_Category=as.factor(MetaData$SexSel_Category)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Postmating competition"))
Model_REML_by_SexSelCat = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelCat)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
1.0179 -2.0359 17.9641 41.5312 21.2477
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0198 0.1408 51 no Study_ID no
sigma^2.2 0.0236 0.1537 85 no Index no
sigma^2.3 0.0153 0.1237 9 no Class yes
Test for Residual Heterogeneity:
QE(df = 78) = 765.4567, p-val < .0001
Test of Moderators (coefficients 2:7):
QM(df = 6) = 73.6003, p-val < .0001
Model Results:
estimate se zval pval
intrcpt -0.2144 0.1185 -1.8089 0.0705
factor(SexSel_Category)Density 0.4286 0.1396 3.0693 0.0021
factor(SexSel_Category)Mating system 0.5918 0.1040 5.6922 <.0001
factor(SexSel_Category)OSR 0.8227 0.1472 5.5885 <.0001
factor(SexSel_Category)Other 0.5517 0.1194 4.6193 <.0001
factor(SexSel_Category)Premating competition 0.5686 0.1042 5.4557 <.0001
factor(SexSel_Category)Trait-based 0.1550 0.1167 1.3280 0.1842
ci.lb ci.ub
intrcpt -0.4466 0.0179 .
factor(SexSel_Category)Density 0.1549 0.7023 **
factor(SexSel_Category)Mating system 0.3881 0.7956 ***
factor(SexSel_Category)OSR 0.5342 1.1113 ***
factor(SexSel_Category)Other 0.3176 0.7858 ***
factor(SexSel_Category)Premating competition 0.3643 0.7728 ***
factor(SexSel_Category)Trait-based -0.0737 0.3837
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Trait-based"))
Model_REML_by_SexSelCat2 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelCat2)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Density"))
Model_REML_by_SexSelCat3 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelCat3)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Premating competition"))
Model_REML_by_SexSelCat4 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelCat4)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Mating system"))
Model_REML_by_SexSelCat5 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelCat5)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("OSR"))
Model_REML_by_SexSelCat6 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelCat6)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Other"))
Model_REML_by_SexSelCat7 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelCat7)Finally, we computed FDR corrected p-values:
tab2=as.data.frame(round(p.adjust(c(0.0705, 0.5745, 0.1057, 0.0002, .0001, .0001, 0.0011), method = 'fdr'),digit=3),row.names=cbind("Postmating competition","Trait-based","Density",'Premating competition',"Mating system","OSR","Other"))
colnames(tab2)<-cbind('P-value')
tab2 P-value
Postmating competition 0.099
Trait-based 0.575
Density 0.123
Premating competition 0.000
Mating system 0.000
OSR 0.000
Other 0.002Plot sexual selection category (Figure S1)
Here we plot the sexual selection category moderator:
MetaData$SexSel_Category=factor(MetaData$SexSel_Category, levels = rev(c( "Postmating competition","Trait-based" ,"Density","Premating competition" ,"Mating system" , "OSR", "Other")))
ggplot(MetaData, aes(x=SexSel_Category, y=r, fill = SexSel_Category, colour = SexSel_Category)) +
geom_hline(yintercept=0, linetype="longdash", color = "black", linewidth=1)+
geom_flat_violin(position = position_nudge(x = 0.25, y = 0),adjust =1, trim = F,alpha=0.6)+
geom_point(position = position_jitter(width = .1), size = 2.5,alpha=0.6,stroke=0,shape=19)+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat$b[1,1], x=7.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat2$b[1,1], x=6.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat3$b[1,1], x=5.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat4$b[1,1], x=4.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat5$b[1,1], x=3.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat6$b[1,1], x=2.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat7$b[1,1], x=1.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat$ci.lb[1], x=7.25, xend= 7.25, yend= Model_REML_by_SexSelCat$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat2$ci.lb[1], x=6.25, xend= 6.25, yend= Model_REML_by_SexSelCat2$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat3$ci.lb[1], x=5.25, xend= 5.25, yend= Model_REML_by_SexSelCat3$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat4$ci.lb[1], x=4.25, xend= 4.25, yend= Model_REML_by_SexSelCat4$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat5$ci.lb[1], x=3.25, xend= 3.25, yend= Model_REML_by_SexSelCat5$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat6$ci.lb[1], x=2.25, xend= 2.25, yend= Model_REML_by_SexSelCat6$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat7$ci.lb[1], x=1.25, xend= 1.25, yend= Model_REML_by_SexSelCat7$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
ylab(expression(paste("Effect size (", italic("r"),')')))+xlab('Sexual selection category')+coord_flip()+guides(fill = FALSE, colour = FALSE) +
scale_color_manual(values =colpal2)+
scale_fill_manual(values =colpal2)+
scale_x_discrete(labels=rev(c( "Post-mating\ncompetition","Trait-based" ,"Density","Pre-mating\ncompetition" ,"Mating\nsystem" , "OSR", "Other")),expand=c(.1,0))+
annotate("text", x=7, y=1.2, label= "n = 9",size=4.5) +
annotate("text", x=6, y=1.2, label= "n = 12",size=4.5) +
annotate("text", x=5, y=1.2, label= "n = 5",size=4.5) +
annotate("text", x=4, y=1.2, label= "n = 18",size=4.5) +
annotate("text", x=3, y=1.2, label= "n = 23",size=4.5) +
annotate("text", x=2, y=1.2, label= "n = 6",size=4.5) +
annotate("text", x=1, y=1.2, label= "n = 12",size=4.5) + theme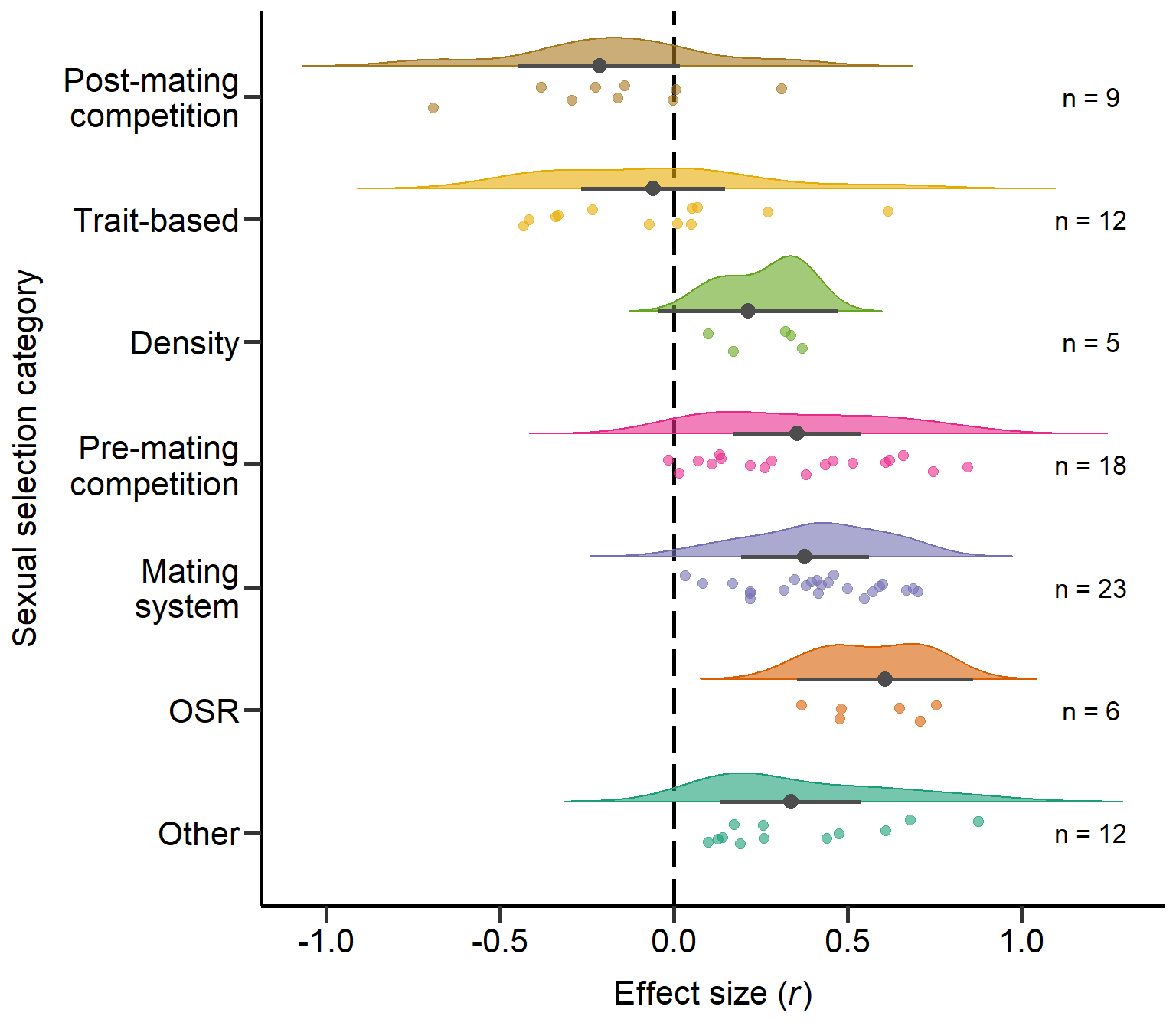
Figure S1: Raincloud plot of correlation coefficients between SSD and different sexual selection categories including sample sizes and estimates with 95%CI from phylogenetic model.
Type of SSD measure
Next we explored the effect of the type of SSD measure (i.e. body mass or size):
MetaData$SSD_Proxy=as.factor(MetaData$SSD_Proxy)
MetaData$SSD_Proxy=relevel(MetaData$SSD_Proxy,c("Body mass"))
Model_REML_by_SSDMeasure = rma.mv(r ~ SSD_Proxy, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SSDMeasure)
Multivariate Meta-Analysis Model (k = 83; method: REML)
logLik Deviance AIC BIC AICc
-23.9277 47.8554 57.8554 69.8276 58.6554
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0277 0.1664 50 no Study_ID no
sigma^2.2 0.0633 0.2516 83 no Index no
sigma^2.3 0.0097 0.0985 9 no Class yes
Test for Residual Heterogeneity:
QE(df = 81) = 1404.5159, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 1.0294, p-val = 0.3103
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.3582 0.1008 3.5543 0.0004 0.1607 0.5557 ***
SSD_ProxyBody size -0.0934 0.0920 -1.0146 0.3103 -0.2737 0.0870
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$SSD_Proxy=relevel(MetaData$SSD_Proxy,c("Body size"))
Model_REML_by_SSDMeasure2 = rma.mv(r ~ SSD_Proxy, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SSDMeasure2)Finally, we computed FDR corrected p-values:
tab3=as.data.frame(round(p.adjust(c(0.0004, 0.0008), method = 'fdr'),digit=3),row.names=cbind("Body mass","Body size"))
colnames(tab3)<-cbind('P-value')
tab3 P-value
Body mass 0.001
Body size 0.001Plot Type of SSD measure (Figure S2A)
Here we plot the type of SSD measure moderator:
MetaData_NAProxy=MetaData[!is.na(MetaData$SSD_Proxy),]
MetaData_NAProxy$SSD_Proxy=as.factor(MetaData_NAProxy$SSD_Proxy)
MetaData_NAProxy$SSD_Proxy=relevel(MetaData_NAProxy$SSD_Proxy,c("Body size"))
ggplot(MetaData_NAProxy, aes(x=SSD_Proxy, y=r, fill = SSD_Proxy, colour = SSD_Proxy)) +
geom_hline(yintercept=0, linetype="longdash", color = "black", linewidth=1)+
geom_flat_violin(position = position_nudge(x = 0.25, y = 0),adjust =1, trim = F,alpha=0.6)+
geom_point(position = position_jitter(width = .1), size = 2.5,alpha=0.6,stroke=0,shape=19)+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SSDMeasure$b[1,1], x=2.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SSDMeasure2$b[1,1], x=1.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SSDMeasure$ci.lb[1], x=2.25, xend= 2.25, yend= Model_REML_by_SSDMeasure$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SSDMeasure2$ci.lb[1], x=1.25, xend= 1.25, yend= Model_REML_by_SSDMeasure2$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
ylab(expression(paste("Effect size (", italic("r"),')')))+xlab('Type of SSD measure')+coord_flip()+guides(fill = FALSE, colour = FALSE) +
scale_color_manual(values =colpal4)+
scale_fill_manual(values =colpal4)+
scale_x_discrete(labels=rev(c("Body mass","Body size")),expand=c(.15,0))+
annotate("text", x=2, y=1.2, label= "n = 37",size=4.5) +
annotate("text", x=1, y=1.2, label= "n = 46",size=4.5) + theme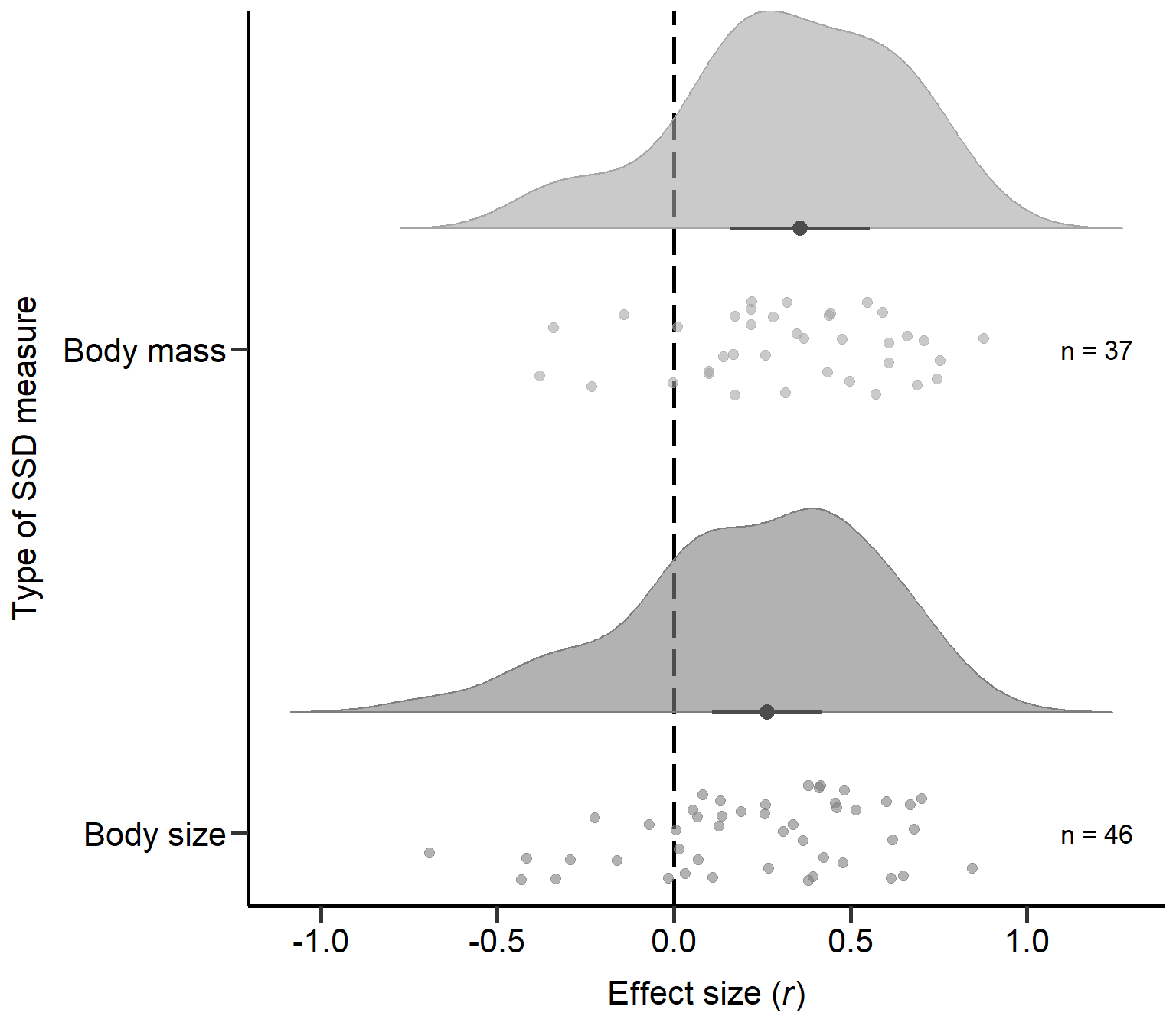
Figure S2A: Raincloud plot of correlation coefficients for different types of SSD measures (i.e. body mass or size) including sample sizes and estimates with 95% CI from phylogenetic model.
SSD measure controlled for body size?
Next we explored the effect if the primary study controlled the SSD for body size (i.e. uncontrolled or controlled):
MetaData$BodySizeControlled=as.factor(MetaData$BodySizeControlled)
MetaData$BodySizeControlled=relevel(MetaData$BodySizeControlled,c("No"))
Model_REML_by_BodySizeCont = rma.mv(r ~ BodySizeControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_BodySizeCont)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
-23.9319 47.8637 57.8637 69.9579 58.6430
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0263 0.1622 51 no Study_ID no
sigma^2.2 0.0619 0.2488 85 no Index no
sigma^2.3 0.0147 0.1214 9 no Class yes
Test for Residual Heterogeneity:
QE(df = 83) = 1445.3959, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 0.2803, p-val = 0.5965
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.3023 0.0885 3.4172 0.0006 0.1289 0.4757 ***
BodySizeControlledYes -0.0546 0.1031 -0.5294 0.5965 -0.2566 0.1474
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$BodySizeControlled=relevel(MetaData$BodySizeControlled,c("Yes"))
Model_REML_by_BodySizeCont2 = rma.mv(r ~ BodySizeControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_BodySizeCont2)Finally, we computed FDR corrected p-values:
tab4=as.data.frame(round(p.adjust(c(0.0006, 0.0337), method = 'fdr'),digit=3),row.names=cbind("uncontrolled","controlled"))
colnames(tab4)<-cbind('P-value')
tab4 P-value
uncontrolled 0.001
controlled 0.034Plot: SSD measure controlled for body size? (Figure S2B)
Here we plot effect sizes if type of SSD measure controlled for body size:
MetaData$BodySizeControlled=as.factor(MetaData$BodySizeControlled)
MetaData$BodySizeControlled=relevel(MetaData$BodySizeControlled,c("Yes"))
ggplot(MetaData, aes(x=BodySizeControlled, y=r, fill = BodySizeControlled, colour = BodySizeControlled)) +
geom_hline(yintercept=0, linetype="longdash", color = "black", linewidth=1)+
geom_flat_violin(position = position_nudge(x = 0.25, y = 0),adjust =1, trim = F,alpha=0.6)+
geom_point(position = position_jitter(width = .1), size = 2.5,alpha=0.6,stroke=0,shape=19)+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_BodySizeCont$b[1,1], x=2.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_BodySizeCont2$b[1,1], x=1.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_BodySizeCont$ci.lb[1], x=2.25, xend= 2.25, yend= Model_REML_by_BodySizeCont$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_BodySizeCont2$ci.lb[1], x=1.25, xend= 1.25, yend= Model_REML_by_BodySizeCont2$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
ylab(expression(paste("Effect size (", italic("r"),')')))+xlab('SSD controlled for body size?')+coord_flip()+guides(fill = FALSE, colour = FALSE) +
scale_color_manual(values =colpal4)+
scale_fill_manual(values =colpal4)+
scale_x_discrete(labels=(c("Controlled","Uncontrolled")),expand=c(.15,0))+
annotate("text", x=2, y=1.2, label= "n = 69",size=4.5) +
annotate("text", x=1, y=1.2, label= "n = 16",size=4.5) + theme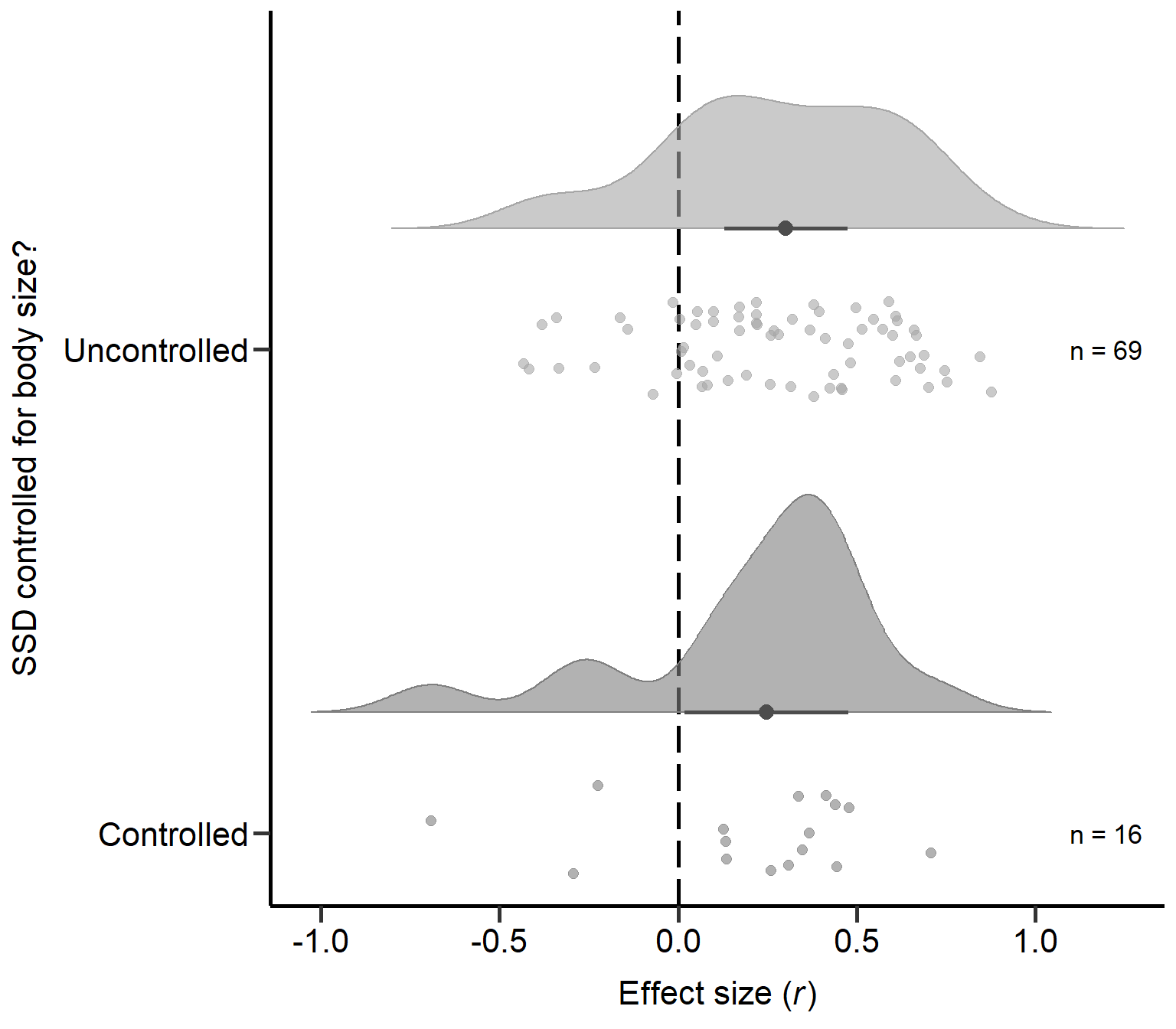
Figure S2B: Raincloud plot of correlation coefficients for primary studies controlling SSD for body size or mass (uncontrolled or controlled) including sample sizes and estimates with 95% CI from phylogenetic model.
Phylogentic vs non-phylogenetic studies
Next we explored the effect of studies controlling for phylogeny vs non-phylogenetic studies:
MetaData$PhyloControlled=as.factor(MetaData$PhyloControlled)
MetaData$PhyloControlled=relevel(MetaData$PhyloControlled,c("No"))
Model_cREML_by_Phylo = rma.mv(r ~ PhyloControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_cREML_by_Phylo)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
-19.4587 38.9175 48.9175 61.0117 49.6967
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0000 0.0000 51 no Study_ID no
sigma^2.2 0.0724 0.2690 85 no Index no
sigma^2.3 0.0203 0.1425 9 no Class yes
Test for Residual Heterogeneity:
QE(df = 83) = 1214.8661, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 11.9815, p-val = 0.0005
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.6301 0.1363 4.6222 <.0001 0.3629 0.8972 ***
PhyloControlledYes -0.3967 0.1146 -3.4614 0.0005 -0.6214 -0.1721 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$PhyloControlled=relevel(MetaData$PhyloControlled,c("Yes"))
Model_cREML_by_Phylo2 = rma.mv(r ~ PhyloControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_cREML_by_Phylo2)Finally, we computed FDR corrected p-values:
tab5=as.data.frame(round(p.adjust(c(0.0001, 0.0096), method = 'fdr'),digit=3),row.names=cbind("Non-phylogenetic","With phylogeny"))
colnames(tab5)<-cbind('P-value')
tab5 P-value
Non-phylogenetic 0.00
With phylogeny 0.01Plot Phylogentic vs non-phylogenetic studies (Figure S2C)
Here we plot the type of SSD measure moderator:
MetaData$PhyloControlled=as.factor(MetaData$PhyloControlled)
MetaData$PhyloControlled=relevel(MetaData$PhyloControlled,c("No"))
ggplot(MetaData, aes(x=PhyloControlled, y=r, fill = PhyloControlled, colour = PhyloControlled)) +
geom_hline(yintercept=0, linetype="longdash", color = "black", linewidth=1)+
geom_flat_violin(position = position_nudge(x = 0.25, y = 0),adjust =1, trim = F,alpha=0.6)+
geom_point(position = position_jitter(width = .1), size = 2.5,alpha=0.6,stroke=0,shape=19)+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Phylo2$b[1,1], x=2.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Phylo$b[1,1], x=1.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Phylo2$ci.lb[1], x=2.25, xend= 2.25, yend= Model_cREML_by_Phylo2$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Phylo$ci.lb[1], x=1.25, xend= 1.25, yend= Model_cREML_by_Phylo$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
ylab(expression(paste("Effect size (", italic("r"),')')))+xlab('Type of analysis')+coord_flip()+guides(fill = FALSE, colour = FALSE) +
scale_color_manual(values =colpal4)+
scale_fill_manual(values =colpal4)+
scale_x_discrete(labels=(c("Phylogenetically \n uncorrected ","Phylogenetically \n corrected ")),expand=c(.15,0))+
annotate("text", x=1, y=1.2, label= "n = 8",size=4.5) +
annotate("text", x=2, y=1.2, label= "n = 77",size=4.5) + theme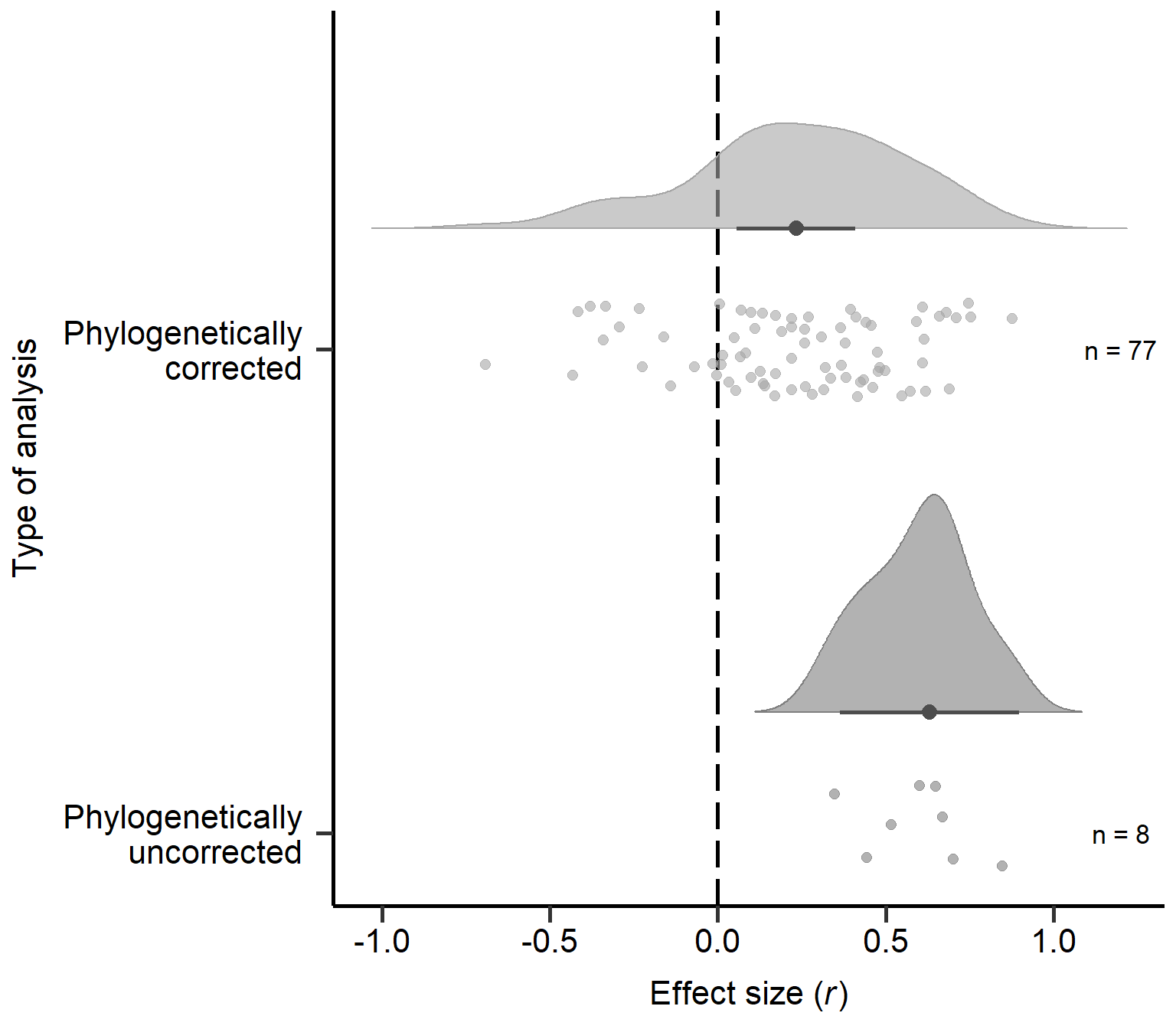
Figure S2C: Raincloud plot of correlation coefficients for non-phylogenetic and phylogenetic analyses in primary studies including sample sizes and estimates with 95% CI from phylogenetic model (Table 4).
Test for publication bias
To test for publication bias, we transformed r into z scores and ran multilevel mixed-effects models (restricted maximum likelihood) with z as the predictor and its standard error as the response with study ID and an observation level random effect. Models were weight by the mean standard error of z across all studies. While the variance in r depends on the effect size and the sample size, the variance in z is only dependent on the sample size. Hence, if z values correlate with the variance in z, this indicates that small studies were only published, if the effect was large, suggesting publication bias.
Model_REML_PublBias = rma.mv(z ~ SE_z, V=rep((mean(SE_z)*mean(SE_z))*N,length(SE_z)), data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML",control=list(rel.tol=1e-8))
summary(Model_REML_PublBias)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
-101.0851 202.1703 212.1703 224.2645 212.9495
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0000 0.0000 51 no Study_ID no
sigma^2.2 0.0000 0.0000 85 no Index no
sigma^2.3 0.0000 0.0003 9 no Class yes
Test for Residual Heterogeneity:
QE(df = 83) = 14.4691, p-val = 1.0000
Test of Moderators (coefficient 2):
QM(df = 1) = 2.8672, p-val = 0.0904
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.0674 0.2533 0.2663 0.7900 -0.4290 0.5639
SE_z 1.5228 0.8993 1.6933 0.0904 -0.2398 3.2854 .
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Figure S2D
# Extract model predictions
model_p2 <- predict(Model_REML_PublBias)
ggplot(MetaData, aes(x=SE_z, y=z)) +
theme(panel.grid.major = element_blank(), panel.grid.minor = element_blank(),panel.background = element_blank(), axis.line = element_line(colour = "black"))+
theme(axis.text=element_text(size=13),
axis.title=element_text(size=14))+ theme(legend.position="none")+ylab(expression(paste("Effect size (", italic("z"),')')))+
geom_point(shape=16, size = 3,alpha=0.5)+xlab('Standard error')+
geom_hline(yintercept=0, linetype="dashed", color = "black", linewidth=1)+
geom_line( aes(y = model_p2$pred), size = 1)+
geom_ribbon( aes(ymin = model_p2$ci.lb, ymax = model_p2$ci.ub, color = 'black',linetype=NA), alpha = .15,show.legend = F, outline.type = "both") +
theme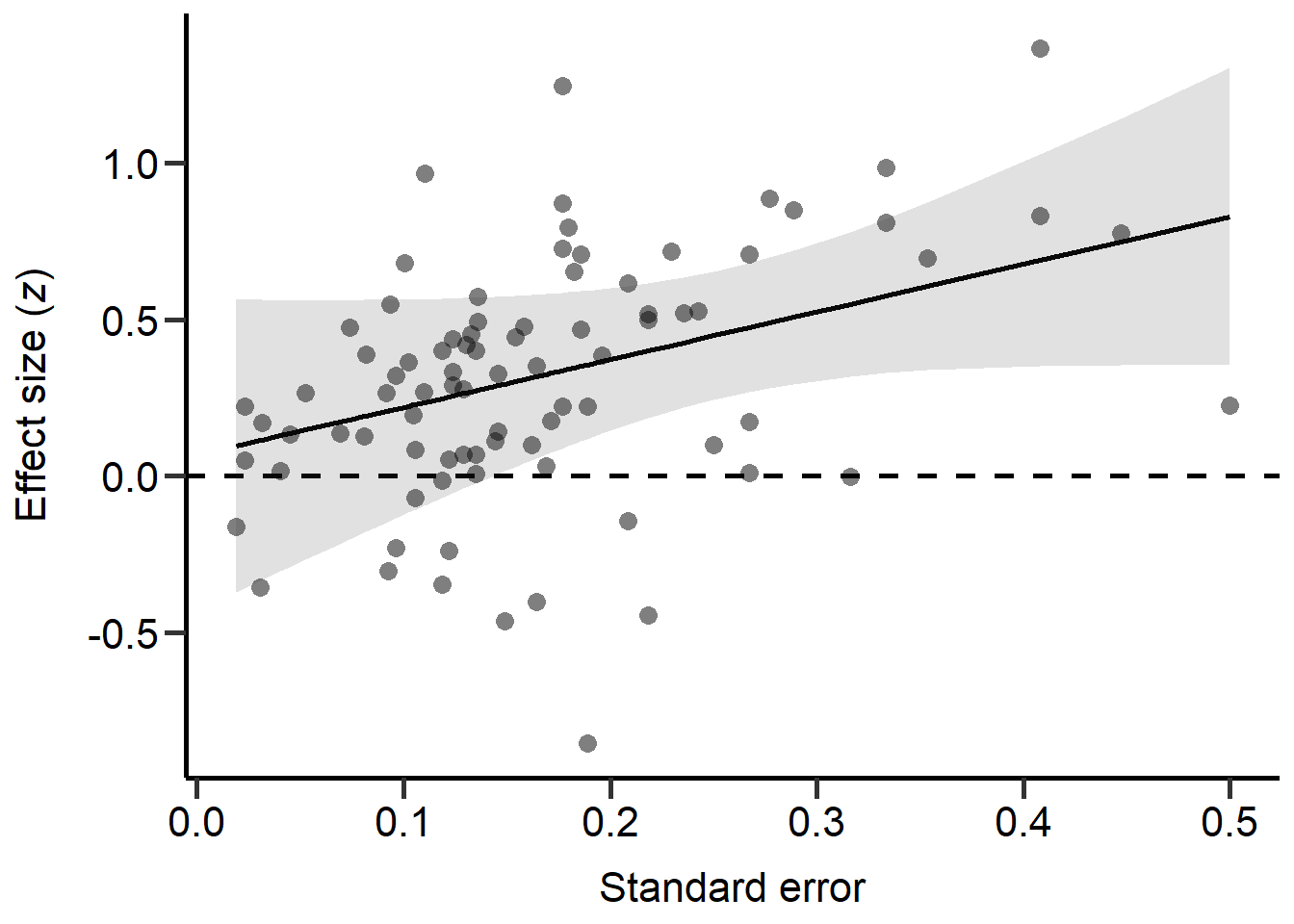
Figure S2D: Scatter plot of the standard error in z scores of each study against the z score of each primary study (transformed correlation coefficients). Dashed line marks a correlation coefficient of zero, black line represents predictions from REML model (controlling for phylogeny) and grey area represents 95% CI on model predictions.
Publication year
Next we explored the effect of the publication year of each study:
Model_REML_by_Year = rma.mv(r ~ Year, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_Year)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
-21.5905 43.1810 53.1810 65.2752 53.9602
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0160 0.1266 51 no Study_ID no
sigma^2.2 0.0624 0.2498 85 no Index no
sigma^2.3 0.0206 0.1435 9 no Class yes
Test for Residual Heterogeneity:
QE(df = 83) = 1136.4440, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 5.5451, p-val = 0.0185
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 17.0962 7.1392 2.3947 0.0166 3.1037 31.0888 *
Year -0.0084 0.0036 -2.3548 0.0185 -0.0153 -0.0014 *
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Plot publication year (Figure S2E)
Here we plot the publication year:
# Extract model predictions
Model_REML_by_Year = rma.mv(r ~ Year, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
model_p4 <- predict(Model_REML_by_Year)
ggplot(MetaData, aes(x=as.numeric(Year), y=r)) +
theme(panel.grid.major = element_blank(), panel.grid.minor = element_blank(),panel.background = element_blank(), axis.line = element_line(colour = "black"))+
theme(axis.text=element_text(size=13),
axis.title=element_text(size=14))+ theme(legend.position="none")+
geom_point(shape=16, size = 3,alpha=0.5)+xlab('Publication year')+ylab(expression(paste("Effect size (", italic("r"),')')))+
geom_hline(yintercept=0, linetype="dashed", color = "black", linewidth=1)+
geom_line( aes(y = model_p4$pred), size = 1)+
geom_ribbon( aes(ymin = model_p4$ci.lb, ymax = model_p4$ci.ub, color = 'black',linetype=NA), alpha = .15,show.legend = F, outline.type = "both") +
theme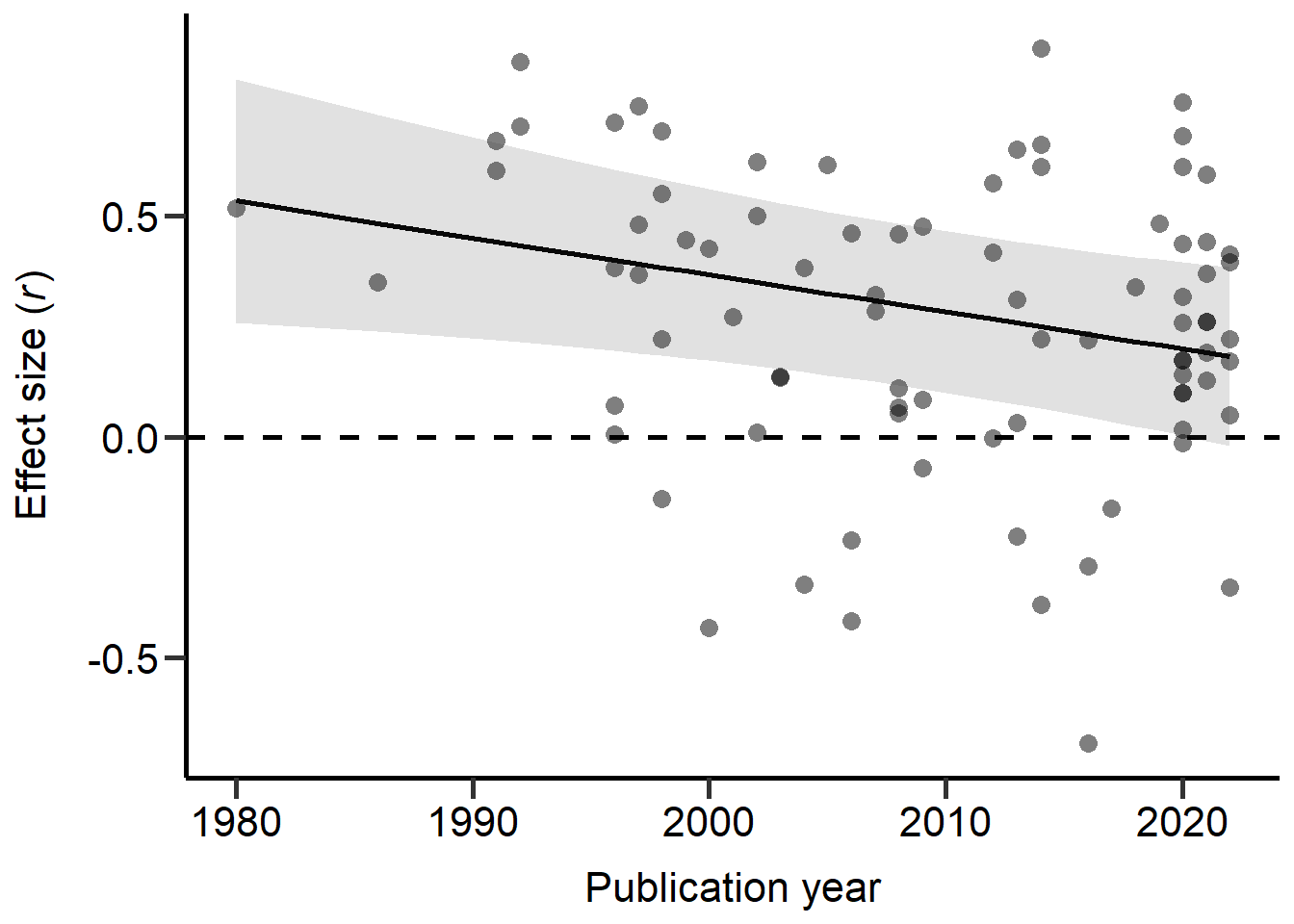
Figure S2E: Scatter plot of correlation coefficients against the publication year of each study. Dashed line marks a correlation coefficient of zero, black line represents predictions from REML model (controlling for phylogeny) and grey area represents 95% CI on model predictions.
Moderator tests for non-phylogenetic models
Here we ran all models without the phylogeny.
Sexual selection mode
The first model explores the effect of the sexual selection mode (i.e. pre-copulatory, post-copulatory or both):
MetaData$SexSel_Mode=relevel(MetaData$SexSel_Mode,c("pre-copulatory"))
Model_REML_by_cSexSelMode = rma.mv(r ~ factor(SexSel_Mode), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelMode)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
-13.2317 26.4634 36.4634 48.4970 37.2529
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0225 0.1501 51 no Study_ID
sigma^2.2 0.0471 0.2171 85 no Index
Test for Residual Heterogeneity:
QE(df = 82) = 1230.9203, p-val < .0001
Test of Moderators (coefficients 2:3):
QM(df = 2) = 27.7973, p-val < .0001
Model Results:
estimate se zval pval ci.lb
intrcpt 0.2592 0.0492 5.2684 <.0001 0.1628
factor(SexSel_Mode)both 0.1603 0.0683 2.3468 0.0189 0.0264
factor(SexSel_Mode)post-copulatory -0.4444 0.1135 -3.9136 <.0001 -0.6669
ci.ub
intrcpt 0.3556 ***
factor(SexSel_Mode)both 0.2942 *
factor(SexSel_Mode)post-copulatory -0.2218 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$SexSel_Mode=relevel(MetaData$SexSel_Mode,c("post-copulatory"))
Model_REML_by_cSexSelMode2 = rma.mv(r ~ factor(SexSel_Mode), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelMode2)
MetaData$SexSel_Mode=relevel(MetaData$SexSel_Mode,c("both"))
Model_REML_by_cSexSelMode3 = rma.mv(r ~ factor(SexSel_Mode), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelMode3)Finally, we computed FDR corrected p-values:
tab1=as.data.frame(round(p.adjust(c(0.0001, 0.0756, .0001), method = 'fdr'),digit=3),row.names=cbind("Pre-copulatory","Post-copulatory","Both"))
colnames(tab1)<-cbind('P-value')
tab1 P-value
Pre-copulatory 0.000
Post-copulatory 0.076
Both 0.000Sexual selection category
Next we explored the effect of the sexual selection category (i.e. density, mating system, operational sex ratio (OSR), post-mating competition, pre-mating competition, trait-based, other):
MetaData$SexSel_Category=as.factor(MetaData$SexSel_Category)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Postmating competition"))
Model_REML_by_cSexSelCat = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelCat)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
0.1558 -0.3116 17.6884 38.8988 20.3355
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0250 0.1582 51 no Study_ID
sigma^2.2 0.0247 0.1571 85 no Index
Test for Residual Heterogeneity:
QE(df = 78) = 765.4567, p-val < .0001
Test of Moderators (coefficients 2:7):
QM(df = 6) = 73.2651, p-val < .0001
Model Results:
estimate se zval pval
intrcpt -0.1685 0.0925 -1.8218 0.0685
factor(SexSel_Category)Other 0.5532 0.1212 4.5647 <.0001
factor(SexSel_Category)OSR 0.8048 0.1462 5.5046 <.0001
factor(SexSel_Category)Mating system 0.5855 0.1043 5.6126 <.0001
factor(SexSel_Category)Premating competition 0.5553 0.1072 5.1809 <.0001
factor(SexSel_Category)Density 0.4346 0.1428 3.0429 0.0023
factor(SexSel_Category)Trait-based 0.1317 0.1154 1.1418 0.2535
ci.lb ci.ub
intrcpt -0.3498 0.0128 .
factor(SexSel_Category)Other 0.3156 0.7907 ***
factor(SexSel_Category)OSR 0.5183 1.0914 ***
factor(SexSel_Category)Mating system 0.3810 0.7899 ***
factor(SexSel_Category)Premating competition 0.3452 0.7653 ***
factor(SexSel_Category)Density 0.1547 0.7146 **
factor(SexSel_Category)Trait-based -0.0944 0.3578
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Trait-based"))
Model_REML_by_cSexSelCat2 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelCat2)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Density"))
Model_REML_by_cSexSelCat3 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelCat3)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Premating competition"))
Model_REML_by_cSexSelCat4 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelCat4)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Mating system"))
Model_REML_by_cSexSelCat5 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelCat5)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("OSR"))
Model_REML_by_cSexSelCat6 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelCat6)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Other"))
Model_REML_by_cSexSelCat7 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelCat7)Finally, we computed FDR corrected p-values:
tab2=as.data.frame(round(p.adjust(c(0.0685, 0.5996, 0.0151, .0001, .0001, .0001, .0001), method = 'fdr'),digit=3),row.names=cbind("Postmating competition","Trait-based","Density",'Premating competition',"Mating system","OSR","Other"))
colnames(tab2)<-cbind('P-value')
tab2 P-value
Postmating competition 0.080
Trait-based 0.600
Density 0.021
Premating competition 0.000
Mating system 0.000
OSR 0.000
Other 0.000Phylogenetic classes
Next we explored the effect of the phylogenetic classes:
MetaData$Class=as.factor(MetaData$Class)
MetaData$Class=relevel(MetaData$Class,c("Nematoda"))
Model_cREML_by_Class = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
-20.3784 40.7568 62.7568 88.3949 66.8818
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0289 0.1701 51 no Study_ID
sigma^2.2 0.0615 0.2480 85 no Index
Test for Residual Heterogeneity:
QE(df = 76) = 1105.0314, p-val < .0001
Test of Moderators (coefficients 2:9):
QM(df = 8) = 11.1317, p-val = 0.1943
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.4221 0.2716 1.5538 0.1202 -0.1103 0.9545
ClassActinopterygii -0.2496 0.3112 -0.8022 0.4225 -0.8594 0.3603
ClassAmphibia -0.2907 0.3233 -0.8992 0.3685 -0.9243 0.3429
ClassAnimalia -0.1962 0.3722 -0.5271 0.5981 -0.9257 0.5333
ClassAves -0.2221 0.2807 -0.7912 0.4288 -0.7724 0.3281
ClassInsecta 0.0288 0.3069 0.0939 0.9252 -0.5727 0.6304
ClassMammalia -0.0056 0.2799 -0.0200 0.9840 -0.5542 0.5430
ClassPisces -0.1638 0.4082 -0.4012 0.6883 -0.9639 0.6363
ClassReptilia -0.3315 0.2959 -1.1203 0.2626 -0.9115 0.2485
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$Class=relevel(MetaData$Class,c("Insecta"))
Model_cREML_by_Class2 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class2)
MetaData$Class=relevel(MetaData$Class,c("Aves"))
Model_cREML_by_Class3 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class3)
MetaData$Class=relevel(MetaData$Class,c("Reptilia"))
Model_cREML_by_Class4 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class4)
MetaData$Class=relevel(MetaData$Class,c("Mammalia"))
Model_cREML_by_Class5 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class5)
MetaData$Class=relevel(MetaData$Class,c("Amphibia"))
Model_cREML_by_Class6 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class6)
MetaData$Class=relevel(MetaData$Class,c("Actinopterygii"))
Model_cREML_by_Class7 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class7)
MetaData$Class=relevel(MetaData$Class,c("Pisces"))
Model_cREML_by_Class8 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class8)
MetaData$Class=relevel(MetaData$Class,c("Animalia"))
Model_cREML_by_Class9 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class9)Finally, we computed FDR corrected p-values:
tab2=as.data.frame(round(p.adjust(c(0.2557 ,0.4534 , 0.3747 ,0.0048 ,0.0016 ,.0001 ,0.1202 , 0.3966 ,0.4405 ), method = 'fdr'),digit=3),row.names=cbind( "Nematoda","Insecta","Aves","Reptilia","Mammalia","Amphibia","Actinopterygii","Pisces","Animalia"))
colnames(tab2)<-cbind('P-value')
tab2 P-value
Nematoda 0.453
Insecta 0.453
Aves 0.453
Reptilia 0.014
Mammalia 0.007
Amphibia 0.001
Actinopterygii 0.270
Pisces 0.453
Animalia 0.453Plot phylogenetic classes (Figure 3)
Here we plot the phylogenetic classes:
MetaData$Class=as.factor(MetaData$Class)
MetaData$Class=factor(MetaData$Class, levels = (c("Animalia","Pisces" ,"Actinopterygii","Amphibia","Mammalia","Reptilia","Aves","Insecta","Nematoda")))
ggplot(MetaData, aes(x=Class, y=r, fill = Class, colour = Class)) +
geom_hline(yintercept=0, linetype="longdash", color = "black", linewidth=1)+
geom_point(position = position_jitter(width = .1), size = 2.5,alpha=0.8,stroke=0,shape=19)+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class$b[1,1], x=9.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal3[9])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class2$b[1,1], x=8.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal3[8])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class3$b[1,1], x=7.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal3[7])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class4$b[1,1], x=6.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal3[6])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class5$b[1,1], x=5.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal3[5])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class6$b[1,1], x=4.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal3[4])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class7$b[1,1], x=3.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal3[3])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class8$b[1,1], x=2.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal3[2])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class9$b[1,1], x=1.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal3[1])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class$ci.lb[1], x=9.35, xend= 9.35, yend= Model_cREML_by_Class$ci.ub[1]), alpha=1,linewidth=1,color=colpal3[9])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class2$ci.lb[1], x=8.35, xend= 8.35, yend= Model_cREML_by_Class2$ci.ub[1]), alpha=1,linewidth=1,color=colpal3[8])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class3$ci.lb[1], x=7.35, xend= 7.35, yend= Model_cREML_by_Class3$ci.ub[1]), alpha=1,linewidth=1,color=colpal3[7])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class4$ci.lb[1], x=6.35, xend= 6.35, yend= Model_cREML_by_Class4$ci.ub[1]), alpha=1,linewidth=1,color=colpal3[6])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class5$ci.lb[1], x=5.35, xend= 5.35, yend= Model_cREML_by_Class5$ci.ub[1]), alpha=1,linewidth=1,color=colpal3[5])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class6$ci.lb[1], x=4.35, xend= 4.35, yend= Model_cREML_by_Class6$ci.ub[1]), alpha=1,linewidth=1,color=colpal3[4])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class7$ci.lb[1], x=3.35, xend= 3.35, yend= Model_cREML_by_Class7$ci.ub[1]), alpha=1,linewidth=1,color=colpal3[3])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class8$ci.lb[1], x=2.35, xend= 2.35, yend= Model_cREML_by_Class8$ci.ub[1]), alpha=1,linewidth=1,color=colpal3[2])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class9$ci.lb[1], x=1.35, xend= 1.35, yend= Model_cREML_by_Class9$ci.ub[1]), alpha=1,linewidth=1,color=colpal3[1])+
ylab(expression(paste("Effect size (", italic("r"),')')))+xlab('Class')+coord_flip()+guides(fill = FALSE, colour = FALSE) +
scale_color_manual(values =colpal3)+
scale_fill_manual(values =colpal3)+
scale_x_discrete(labels=(c("Animalia","Pisces" ,"Actinopterygii","Amphibia","Mammalia","Reptilia","Aves","Insecta","Nematoda")),expand=c(.1,0))+
theme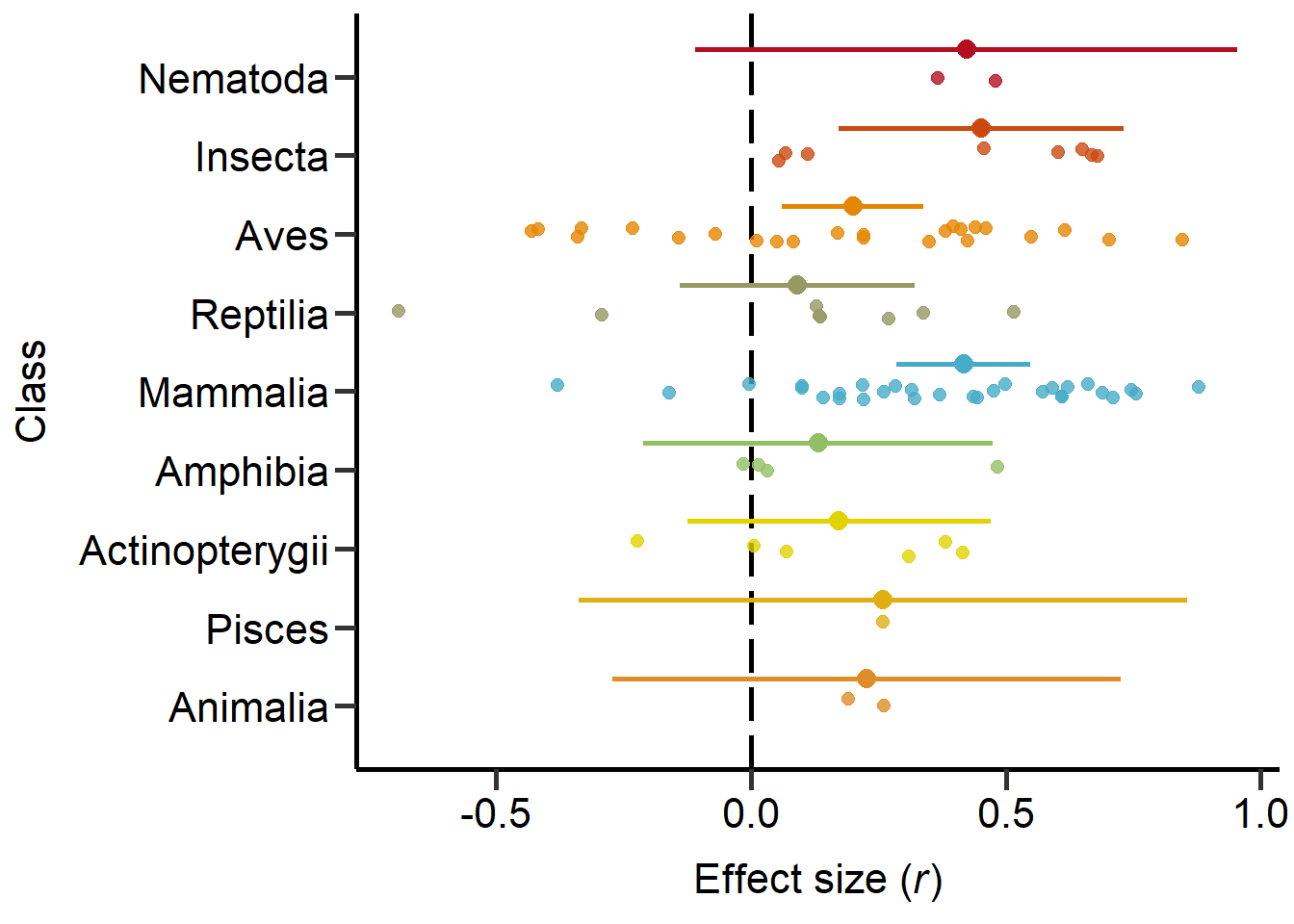
Figure 3: Phylogeny based on classes including sample sizes for the number of studies and number of effect sizes, respectively, and estimates with 95%CI from non-phylogenetic model.
Type of SSD measure
Next we explored the effect of the type of SSD measure (i.e. body mass or size):
MetaData$SSD_Proxy=as.factor(MetaData$SSD_Proxy)
MetaData$SSD_Proxy=relevel(MetaData$SSD_Proxy,c("Body mass"))
Model_REML_by_cSSDMeasure = rma.mv(r ~ SSD_Proxy, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSSDMeasure)
Multivariate Meta-Analysis Model (k = 83; method: REML)
logLik Deviance AIC BIC AICc
-24.2720 48.5441 56.5441 66.1219 57.0704
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0316 0.1777 50 no Study_ID
sigma^2.2 0.0635 0.2521 83 no Index
Test for Residual Heterogeneity:
QE(df = 81) = 1404.5159, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 2.1342, p-val = 0.1440
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.3542 0.0626 5.6615 <.0001 0.2316 0.4768 ***
SSD_ProxyBody size -0.1196 0.0818 -1.4609 0.1440 -0.2800 0.0408
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$SSD_Proxy=relevel(MetaData$SSD_Proxy,c("Body size"))
Model_REML_by_cSSDMeasure2 = rma.mv(r ~ SSD_Proxy, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSSDMeasure2)Finally, we computed FDR corrected p-values:
tab3=as.data.frame(round(p.adjust(c(.0001, .0001), method = 'fdr'),digit=3),row.names=cbind("Body mass","Body size"))
colnames(tab3)<-cbind('P-value')
tab3 P-value
Body mass 0
Body size 0SSD measure controlled for body size?
Next we explored the effect if the primary study controlled the SSD for body size (i.e. uncontrolled or controlled):
MetaData$BodySizeControlled=as.factor(MetaData$BodySizeControlled)
MetaData$BodySizeControlled=relevel(MetaData$BodySizeControlled,c("No"))
Model_REML_by_cBodySizeCont = rma.mv(r ~ BodySizeControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cBodySizeCont)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
-24.7551 49.5102 57.5102 67.1855 58.0230
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0334 0.1828 51 no Study_ID
sigma^2.2 0.0626 0.2502 85 no Index
Test for Residual Heterogeneity:
QE(df = 83) = 1445.3959, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 0.6106, p-val = 0.4346
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.2983 0.0459 6.4948 <.0001 0.2083 0.3883 ***
BodySizeControlledYes -0.0799 0.1023 -0.7814 0.4346 -0.2804 0.1205
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$BodySizeControlled=relevel(MetaData$BodySizeControlled,c("Yes"))
Model_REML_by_cBodySizeCont2 = rma.mv(r ~ BodySizeControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cBodySizeCont2)Finally, we computed FDR corrected p-values:
tab4=as.data.frame(round(p.adjust(c(.0001, 0.0168), method = 'fdr'),digit=3),row.names=cbind("uncontrolled","controlled"))
colnames(tab4)<-cbind('P-value')
tab4 P-value
uncontrolled 0.000
controlled 0.017Phylogentic vs non-phylogenetic studies
Next we explored the effect of studies controlling for phylogeny vs non-phylogenetic studies:
MetaData$PhyloControlled=as.factor(MetaData$PhyloControlled)
MetaData$PhyloControlled=relevel(MetaData$PhyloControlled,c("No"))
Model_cREML_by_cPhylo = rma.mv(r ~ PhyloControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_cPhylo)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
-21.6552 43.3104 51.3104 60.9858 51.8232
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0157 0.1252 51 no Study_ID
sigma^2.2 0.0698 0.2642 85 no Index
Test for Residual Heterogeneity:
QE(df = 83) = 1214.8661, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 7.8737, p-val = 0.0050
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.5883 0.1176 5.0034 <.0001 0.3578 0.8187 ***
PhyloControlledYes -0.3478 0.1240 -2.8060 0.0050 -0.5908 -0.1049 **
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$PhyloControlled=relevel(MetaData$PhyloControlled,c("Yes"))
Model_cREML_by_cPhylo2 = rma.mv(r ~ PhyloControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_cPhylo2)Finally, we computed FDR corrected p-values:
tab5=as.data.frame(round(p.adjust(c(0.0001, 0.0001), method = 'fdr'),digit=3),row.names=cbind("Non-phylogenetic","With phylogeny"))
colnames(tab5)<-cbind('P-value')
tab5 P-value
Non-phylogenetic 0
With phylogeny 0Test for publication bias
Model_cREML_PublBias = rma.mv(z ~ SE_z, V=rep(((mean(SE_z)*mean(SE_z))*N),length(SE_z)), data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_PublBias)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
-101.0851 202.1703 210.1703 219.8456 210.6831
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0000 0.0000 51 no Study_ID
sigma^2.2 0.0000 0.0000 85 no Index
Test for Residual Heterogeneity:
QE(df = 83) = 14.4691, p-val = 1.0000
Test of Moderators (coefficient 2):
QM(df = 1) = 2.8672, p-val = 0.0904
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.0674 0.2533 0.2663 0.7900 -0.4290 0.5638
SE_z 1.5228 0.8993 1.6933 0.0904 -0.2398 3.2854 .
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Publication year
Next we explored the effect of the publication year of each study:
Model_cREML_by_Year = rma.mv(r ~ Year, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Year)
Multivariate Meta-Analysis Model (k = 85; method: REML)
logLik Deviance AIC BIC AICc
-23.2555 46.5110 54.5110 64.1863 55.0238
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0293 0.1712 51 no Study_ID
sigma^2.2 0.0617 0.2484 85 no Index
Test for Residual Heterogeneity:
QE(df = 83) = 1136.4440, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 3.7310, p-val = 0.0534
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 15.0587 7.6506 1.9683 0.0490 0.0639 30.0535 *
Year -0.0074 0.0038 -1.9316 0.0534 -0.0148 0.0001 .
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Forest plot
# Sort data by Class
MetaData$Class=as.factor(MetaData$Class)
MetaData$Class=factor(MetaData$Class, levels = c("Animalia","Pisces" ,"Actinopterygii","Amphibia","Mammalia","Reptilia","Aves","Insecta","Nematoda"))
MetaData_sorted <-MetaData[order(MetaData$Class),]
# Add global effect size to data
forestData=MetaData_sorted[,c(3,5,12,17,20,21)]
GlobalES=as.data.frame(cbind('Global effect size','','Global effect size',Model_REML_Null$b[1,1],Model_REML_Null$ci.lb[1],Model_REML_Null$ci.ub[1]))
colnames(GlobalES)=c('AuthorsAndYear','Class','SexSel_Mode','r','lCI','uCI')
forestData=rbind(GlobalES,forestData)
forestData[,c(4:6)]=lapply(forestData[,c(4:6)],as.numeric)
forestData$SexSel_Mode=as.factor(forestData$SexSel_Mode)
forestData$SexSel_Mode=factor(forestData$SexSel_Mode, levels = c("pre-copulatory","post-copulatory" ,"both","Global effect size"))Figure 1
ggplot(data=forestData, aes(y=1:nrow(forestData), x=r, xmin=lCI, xmax=uCI,color=SexSel_Mode)) +
geom_vline(xintercept=0, color='black', linetype='dashed', alpha=.5,linewidth=0.8) +
geom_point(size = ifelse(1:nrow(forestData) == c(1), 2.75, 1.8))+
geom_errorbarh(height=0,size = ifelse(1:nrow(forestData) == c(1), 1.25, 1))+
scale_y_continuous(breaks=seq(1, nrow(forestData), by=1), labels=forestData$AuthorsAndYear,limits=c(1,length(forestData$AuthorsAndYear)),expand=c(0.015,0.015)) +
labs(title='', x=expression(paste(italic("r "),'(95% CI)')), y = 'Study') +
theme_classic()+theme(axis.text.x=element_text(size=12),
axis.title=element_text(size=12))+ labs(color = c("Sexual selection mode"))+
scale_colour_manual(values=Meta_col, labels=c('Pre-copulatory','Post-copulatory',"Both","Global effect size"))+
guides(color = guide_legend(override.aes = list(size = .9)))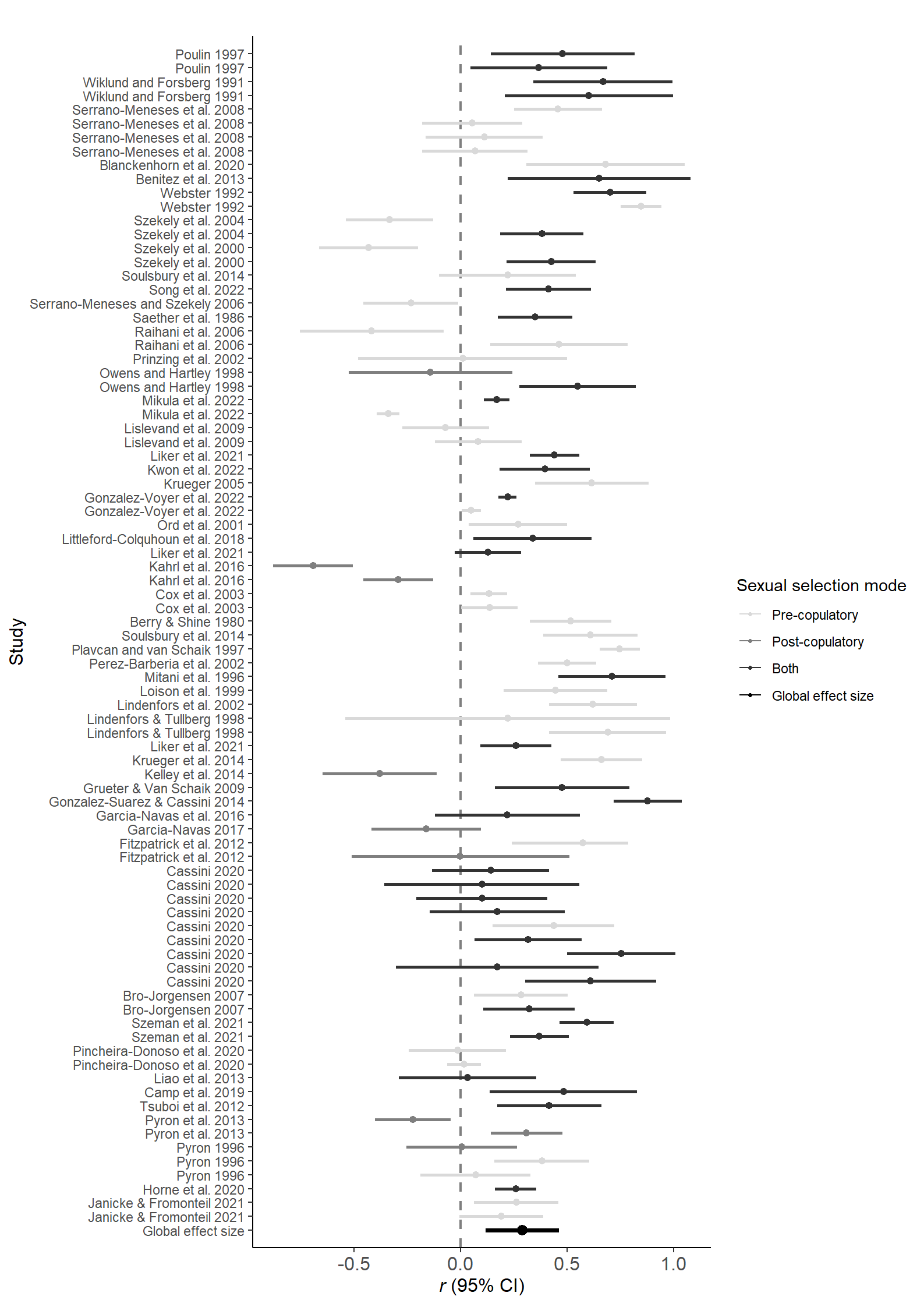
Figure 1: Forest plot including correlation coefficients (±95% CI), as well as the global effect size (in grey) with and without controlling for phylogeny. Different animal classes highlighted by background colors (see Figure 3 for class names) and sexual selection modes pre- (red), post-copulatory (blue) and both (green) by color of effect size.
sessionInfo()R version 4.2.3 (2023-03-15 ucrt)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19045)
Matrix products: default
locale:
[1] LC_COLLATE=German_Germany.utf8 LC_CTYPE=German_Germany.utf8
[3] LC_MONETARY=German_Germany.utf8 LC_NUMERIC=C
[5] LC_TIME=German_Germany.utf8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] wesanderson_0.3.6 PupillometryR_0.0.4 rlang_1.1.0
[4] dplyr_1.1.1 cowplot_1.1.1 ggdist_3.3.0
[7] MCMCglmm_2.34 coda_0.19-4 RColorBrewer_1.1-3
[10] ggplot2_3.4.2 data.table_1.14.8 multcomp_1.4-23
[13] TH.data_1.1-2 survival_3.5-3 mvtnorm_1.1-3
[16] psych_2.3.3 pwr_1.3-0 MASS_7.3-58.2
[19] metafor_4.2-0 numDeriv_2016.8-1.1 metadat_1.2-0
[22] Matrix_1.5-3 matrixcalc_1.0-6 ape_5.7-1
[25] workflowr_1.7.0
loaded via a namespace (and not attached):
[1] httr_1.4.5 sass_0.4.5 jsonlite_1.8.4
[4] splines_4.2.3 bslib_0.4.2 getPass_0.2-2
[7] distributional_0.3.2 highr_0.10 tensorA_0.36.2
[10] yaml_2.3.7 pillar_1.9.0 lattice_0.20-45
[13] glue_1.6.2 digest_0.6.31 promises_1.2.0.1
[16] colorspace_2.1-0 sandwich_3.0-2 htmltools_0.5.5
[19] httpuv_1.6.9 pkgconfig_2.0.3 corpcor_1.6.10
[22] scales_1.2.1 processx_3.8.0 whisker_0.4.1
[25] later_1.3.0 cubature_2.0.4.6 git2r_0.31.0
[28] tibble_3.2.1 generics_0.1.3 farver_2.1.1
[31] cachem_1.0.7 withr_2.5.0 cli_3.6.1
[34] mnormt_2.1.1 magrittr_2.0.3 evaluate_0.20
[37] ps_1.7.3 fs_1.6.1 fansi_1.0.4
[40] nlme_3.1-162 tools_4.2.3 lifecycle_1.0.3
[43] stringr_1.5.0 munsell_0.5.0 callr_3.7.3
[46] compiler_4.2.3 jquerylib_0.1.4 grid_4.2.3
[49] rstudioapi_0.14 labeling_0.4.2 rmarkdown_2.21
[52] gtable_0.3.3 codetools_0.2-19 R6_2.5.1
[55] zoo_1.8-12 knitr_1.42 fastmap_1.1.1
[58] utf8_1.2.3 mathjaxr_1.6-0 rprojroot_2.0.3
[61] stringi_1.7.12 parallel_4.2.3 Rcpp_1.0.10
[64] vctrs_0.6.1 tidyselect_1.2.0 xfun_0.38