Demographic inferences
Emily Baker and Maeva Techer
2025-09-15
Last updated: 2025-09-15
Checks: 6 1
Knit directory:
locust-comparative-genomics/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221025) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version b7729ad. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis/2_signatures-selection_cache/
Ignored: analysis/figure/
Ignored: code/.DS_Store
Ignored: code/scripts/.DS_Store
Ignored: code/scripts/pal2nal.v14/.DS_Store
Ignored: data/.DS_Store
Ignored: data/DEG_results/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/americana/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/cancellata/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/cubense/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/gregaria/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/nitens/.DS_Store
Ignored: data/HYPHY_selection/.DS_Store
Ignored: data/HYPHY_selection/ParsedABSRELResults_unlabeled/.DS_Store
Ignored: data/HYPHY_selection/pathway_enrichment/.DS_Store
Ignored: data/HYPHY_selection/pathway_enrichment/americana/
Ignored: data/HYPHY_selection/pathway_enrichment/cancellata/
Ignored: data/HYPHY_selection/pathway_enrichment/cubense/
Ignored: data/HYPHY_selection/pathway_enrichment/nitens/
Ignored: data/HYPHY_selection/pathway_enrichment/piceifrons/
Ignored: data/WGCNA/.DS_Store
Ignored: data/WGCNA/input/.DS_Store
Ignored: data/WGCNA/input/Bulk_RNAseq/.DS_Store
Ignored: data/WGCNA/output/.DS_Store
Ignored: data/WGCNA/output/Bulk_RNAseq/.DS_Store
Ignored: data/WGCNA/output/Bulk_RNAseq/gregaria/.DS_Store
Ignored: data/behavioral_data/.DS_Store
Ignored: data/behavioral_data/Raw_data/.DS_Store
Ignored: data/cafe5_results/.DS_Store
Ignored: data/list/.DS_Store
Ignored: data/list/Bulk_RNAseq/.DS_Store
Ignored: data/list/GO_Annotations/.DS_Store
Ignored: data/list/GO_Annotations/DesertLocustR/.DS_Store
Ignored: data/list/excluded_loci/.DS_Store
Ignored: data/orthofinder/.DS_Store
Ignored: data/orthofinder/Polyneoptera/.DS_Store
Ignored: data/orthofinder/Polyneoptera/Results_I2_iqtree/.DS_Store
Ignored: data/orthofinder/Polyneoptera/Results_I2_iqtree/Orthogroups/.DS_Store
Ignored: data/orthofinder/Polyneoptera/Results_I2_withDaust/.DS_Store
Ignored: data/orthofinder/Polyneoptera/Results_I2_withDaust/Orthogroups/.DS_Store
Ignored: data/orthofinder/Schistocerca/.DS_Store
Ignored: data/orthofinder/Schistocerca/Results_I2/.DS_Store
Ignored: data/orthofinder/Schistocerca/Results_I2/Orthogroups/.DS_Store
Ignored: data/overlap/.DS_Store
Ignored: data/pathway_enrichment/.DS_Store
Ignored: data/pathway_enrichment/OLD/.DS_Store
Ignored: data/pathway_enrichment/OLD/custom_sgregaria_orgdb/.DS_Store
Ignored: data/pathway_enrichment/REVIGO_results/.DS_Store
Ignored: data/pathway_enrichment/REVIGO_results/BP/.DS_Store
Ignored: data/pathway_enrichment/REVIGO_results/CC/.DS_Store
Ignored: data/pathway_enrichment/REVIGO_results/MF/.DS_Store
Ignored: data/pathway_enrichment/cancellata/.DS_Store
Ignored: data/pathway_enrichment/gregaria/.DS_Store
Ignored: data/pathway_enrichment/nitens/Thorax/
Ignored: data/pathway_enrichment/piceifrons/.DS_Store
Ignored: data/readcounts/.DS_Store
Ignored: data/readcounts/Bulk_RNAseq/.DS_Store
Ignored: data/readcounts/RNAi/.DS_Store
Untracked files:
Untracked: data/RefSeq/
Unstaged changes:
Modified: analysis/2_psmc-analysis.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/2_psmc-analysis.Rmd) and
HTML (docs/2_psmc-analysis.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 6367e32 | Maeva TECHER | 2025-09-04 | Updates PSMC |
| html | 6367e32 | Maeva TECHER | 2025-09-04 | Updates PSMC |
| html | 2b0f55c | Maeva TECHER | 2025-06-05 | Build site. |
| html | 9a03ca6 | Maeva TECHER | 2025-06-05 | Update website |
| html | fb90bdd | Maeva TECHER | 2025-06-05 | Build site. |
| html | 3e696d6 | Maeva TECHER | 2025-06-05 | Adding ortho heatmap |
| html | ff2618d | Maeva TECHER | 2025-06-04 | Build site. |
| html | 68a7e4f | Maeva TECHER | 2025-06-04 | Build site. |
| Rmd | 4e391c3 | Maeva TECHER | 2025-05-30 | add new analysis orthology, synteny |
| Rmd | cacc1db | Maeva TECHER | 2025-05-02 | updates files |
How are locusts and grasshoppers demographic history varying over time?
1. What is PSMC?
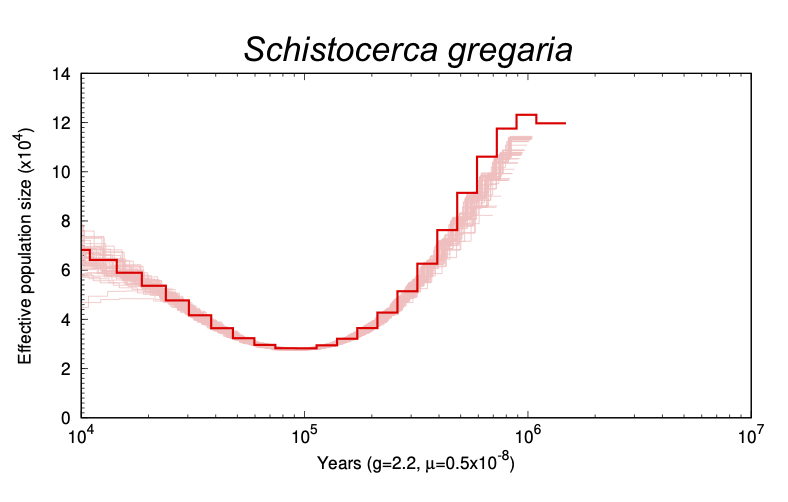
The Pairwise Sequentially Markovian Coalescent Model (PSMC) is a model that infers a species’ population history using high coverage whole genome data (here we will use HiC) from the species in question. This process is motivated by “Coalescence Theory”, which is a statistical theory that relates the size of a population to the coalescence times in the genealogy of sampled individuals (Mather et al.). This is done by mapping HiC reads back to the genome and searching for loci splits via variant calling and bootstrapping results to create a coalescence model.
To have estimates that matters for our Schistocerca species, we must know or approximate to a close relative: 1) the mutation rate and 2) the generation time.
Mutation rate: As we do not know the exact mutation rate for Orthoptera and more particularly for Schistocerca, we will use the mutation rate for the model insect Drosophila melanogaster which was estimated around 2.8 × 10−9 per site per generation Keightley et al., 2013.
Generation time: This parameter is a bit more tricky as the number of generations varies between field vs lab populations and between species. In the preprint by Chapuis et al., 2020, the average generation time for S. gregaria is estimated to be 3 generations per year and in optimal weather conditions, it can have up to 5 generations on the field leading to a plague. But in the poor weather conditions, solitarious populations probably go through 2 generations.
For other species, our expert team observed the following: S. piceifrons usually has two generations per year, one with a reproductive diapause and one without. It may have up to three, but it is a rare occurence. As for S. cancellata, 2 generations per year seems to be what is observed in normal conditions, but in outbreak years, it may have more.
For non-swarming species, S. americana probably has 2 generations per year but it’s not really synchronized and it depends on where it lives. In Florida it can be found year around, but in Texas, we observed around 2 generations per year. For S. serialis cubense, we observed two generations per year but this is based on sparse observations. S. nitens has one generation per year due to a long diapause.
So we decided to plot the main graphs with the following
approximations:
- 2 generations per year (g = 0.5) for locusts and non-swarming species
S. americana and S. serialis cubense. - 2 generations
per year (g = 1) for the sedentary species S. nitens.
2. Installation & Running
In the drop down below the full script to run the entire pipeline in one slurm script is available. Below this, each step is broken down for informational and troubleshooting purposes. You can run this procedure step-by-step or all in one script, but for the first run I would recommend doing a step-by-step run as it simplifies troubleshooting and familiarizes you with the procedure.
Below we are showing the script for S. gregaria which was
replicated for all species.
Download HiC Data from
NCBI
1. First, we must map the Hi-C reads back to the original genome using BWA. Doing this step on the (enormous) S. gregaria genome (8.3 Gb) took around 1-2 days. We do this using the following procedure:
#!/bin/bash
##NECESSARY JOB SPECIFICATIONS
#SBATCH --job-name=create-unsorted-alignment #Set the job name to "psmc-referencegenome"
#SBATCH --time=3-00:00:00 #Set the wall clock limit to 2 hours
#SBATCH --ntasks=1 #Request 1 task
#SBATCH --cpus-per-task=8 #Request 8 cpus
#SBATCH --mem=30G #Request 30GB per node
#SBATCH --output=stdout.%x.%j
#SBATCH --error=stderr.%x.%j
# Align with BWA
module purge
ml GCC/13.2.0 BWA/0.7.18 SAMtools/1.19.2
# bwa index -p Sgregaria GCF_023897955.1_iqSchGreg1.2_genomic.fa
# assuming you have 8 threads, not that this .sam file will be huge, and we'll remove it at the end
bwa mem -t 8 Sgregaria SRR17149718_1.fastq SRR17149718_2.fastq > SRR17149718.sam
# convert .sam to .bam
samtools view -S -b SRR17149718.sam > unsorted_alignment.bam
2. Sort the unsorted .bam file
##NECESSARY JOB SPECIFICATIONS
#SBATCH --job-name=sort-alignment #Set the job name to "psmc-referencegenome"
#SBATCH --time=1-00:00:00 #Set the wall clock limit to 2 hours
#SBATCH --ntasks=1 #Request 1 task
#SBATCH --cpus-per-task=8 #Request 8 cpus
#SBATCH --mem=30G #Request 30GB per node
#SBATCH --output=stdout.%x.%j
#SBATCH --error=stderr.%x.%j
# sort the .bam file
module purge
module load picard/2.25.1-Java-11
java -jar $EBROOTPICARD/picard.jar SortSam \
INPUT=unsorted_alignment.bam \
OUTPUT=sorted.bam \
SORT_ORDER=coordinate
3. Generate a variant consensus file from the sorted .bam file
#!/bin/bash
##NECESSARY JOB SPECIFICATIONS
#SBATCH --job-name=create-var-con # Set the job name to "create-var-con"
#SBATCH --time=2-00:00:00 # Set the wall clock limit to 2 days
#SBATCH --ntasks=1 # Request 1 task
#SBATCH --cpus-per-task=8 # Request 8 cpus
#SBATCH --mem=30G # Request 30GB per node
#SBATCH --mail-type=ALL # Email updates for all steps
#SBATCH --mail-user=user@mail.edu # Email to this address
#SBATCH --output=stdout.%x.%j # Name stdout file after job name and ID
#SBATCH --error=stderr.%x.%j # Name stderr file after job name and ID
module purge
ml GCC/13.2.0 BCFtools/1.19
# Generate consensus fastq for PSMC
bcftools mpileup -Q 20 -q 20 -f GCF_023897955.1_iqSchGreg1.2_genomic.fa \
sorted.bam --threads 8 | \
bcftools call -c --threads 8 | \
vcfutils.pl vcf2fq -d 1 -D 100 -Q 20 > variant_consensus.fq4. Generate PSMC results
#!/bin/bash
##NECESSARY JOB SPECIFICATIONS
#SBATCH --job-name=psmc-input-and-plot
#SBATCH --time=4-00:00:00
#SBATCH --ntasks=32
#SBATCH --cpus-per-task=8
#SBATCH --mem=120G
#SBATCH --mail-type=ALL
#SBATCH --mail-user=ecbaker7@tamu.edu
#SBATCH --output=stdout.%x.%j
#SBATCH --error=stderr.%x.%j
module purge
ml GCC/13.2.0 psmc/0.6.5_20221121 parallel/20240322 gnuplot/6.0.1 texlive/20230313
ml Ghostscript/10.02.1
# Convert .fq to .psmcfa
fq2psmcfa variant_consensus.fq > psmc_input.psmcfa
# Generate PSMC results
psmc -N25 -t15 -r5 -p "4+25*2+4+6" -o sample.psmc psmc_input.psmcfa
mkdir boot
# Prepare bootstrap input
splitfa psmc_input.psmcfa > psmc_input.split.psmcfa
# Bootstrap results and plot
seq 100 | parallel -j ${SLURM_CPUS_PER_TASK:-8} \
'psmc -N25 -t15 -r5 -b -p "4+25*2+4+6" \
-o boot100/sample_r{}.psmc psmc_input.split.psmcfa'
cat sample.psmc boot/sample_r*.psmc > sample_combined.psmc
# Combine for plotting
cat sample.psmc boot/sample_r*.psmc > sample_combined100.psmc
# plotting considering 2 generations per year
psmc_plot.pl -p -g 0.5 -u 2.8e-9 bootstrapped_plot100 sample_combined100.psmc3. Plot multiple species
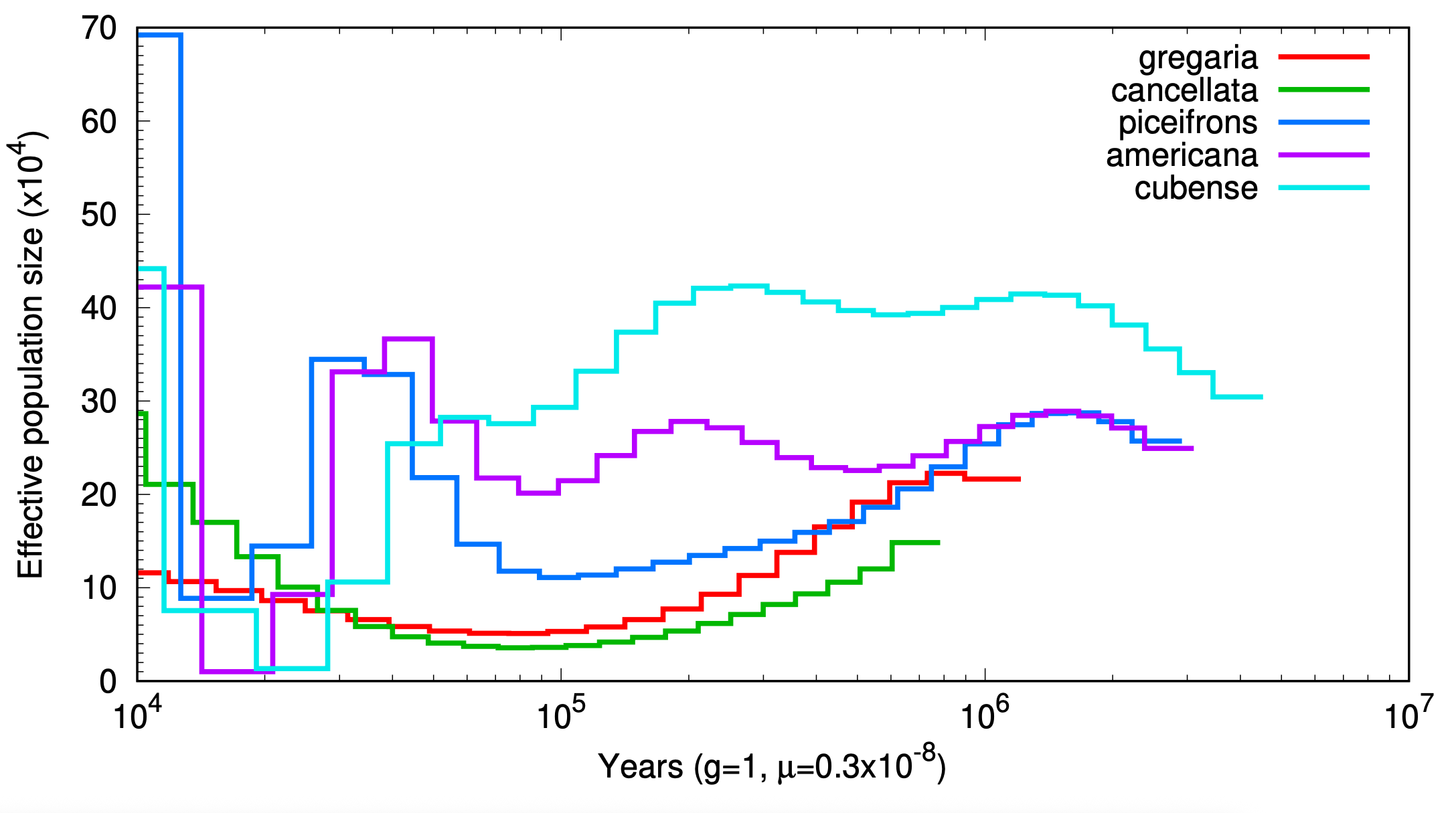
Here if we want to plot for multiple species, we can call the main line as follow:
labels="gregaria,cancellata,piceifrons,americana,cubense"
# 1 generation / year
psmc_plot.pl -p -w 5 -g 1 -u 2.8e-9 -M "$labels" allsp_g1 greg/sample.psmc canc/sample.psmc pice/sample.psmc amer/sample.psmc seri/sample.psmc
# 2 generations / year
psmc_plot.pl -p -w 5 -g 0.5 -u 2.8e-9 -M "$labels" allsp_g0.5 greg/sample.psmc canc/sample.psmc pice/sample.psmc amer/sample.psmc seri/sample.psmc
# 3 generations / year
psmc_plot.pl -p -w 5 -g 0.33 -u 2.8e-9 -M "$labels" allsp_g0.33 greg/sample.psmc canc/sample.psmc pice/sample.psmc amer/sample.psmc seri/sample.psmc
# 4 generations / year
psmc_plot.pl -p -w 5 -g 0.25 -u 2.8e-9 -M "$labels" allsp_g0.25 greg/sample.psmc canc/sample.psmc pice/sample.psmc amer/sample.psmc seri/sample.psmc
# 5 generations / year
psmc_plot.pl -p -w 5 -g 0.2 -u 2.8e-9 -M "$labels" allsp_g0.2 greg/sample.psmc canc/sample.psmc pice/sample.psmc amer/sample.psmc seri/sample.psmc
If one wants to add the bootstraps with it, we recommend to export
individually the plot using the sample_combined100.psmc and
-Y as a limit for scale.
OPTIONAL: Adding a title to your plot
There is no option to add a plot title in the psmc module, so if you want your plot to be named, you’ll have to directly edit the gnuplot file. Search for a file in your PSMC dirctory ending in the extension .gnuplot or .gp. Then type
nano YOURFILE.gpinto your terminal to edit the gnuplot file. Then, find where the x and y labels are set. The file layout should look something like this:
set size 1, 0.8;
set xran [10000:*];
set log x;
set format x "10^{%L}";
set mxtics 10;
set mytics 10;
unset grid;
set key off;
set xtics font "Helvetica,16";
set ytics nomirror font "Helvetica,16";
set xlab "Years (g=2.2, {/Symbol m}=0.5x10^{-8})" font "Helvetica,16";
set t po eps enhance so co "Helvetica,16";
set yran [0:*];
set ylab "Effective population size (x10^4)" font "Helvetica,16";
set out "Schistocerca piceifrons.eps";
set style line 1 lt 1 lc rgb "#FF0000" lw 4;and add the line set title “Your Title” below the set x and y label commands. For example, my file looks like this for Schistocerca piceifrons:
set size 1, 0.8;
set xran [10000:*];
set log x;
set format x "10^{%L}";
set mxtics 10;
set mytics 10;
unset grid;
set key off;
set xtics font "Helvetica,16";
set ytics nomirror font "Helvetica,16";
set xlab "Years (g=2.2, {/Symbol m}=0.5x10^{-8})" font "Helvetica,16";
set t po eps enhance so co "Helvetica,16";
set yran [0:*];
set ylab "Effective population size (x10^4)" font "Helvetica,16";
set title "Schistocerca piceifrons" font "Helvetica-Italic,24";
set out "Schistocerca piceifrons.eps";
set style line 1 lt 1 lc rgb "#FF0000" lw 4;Citations
Mather, Niklas, et al. “A Practical Introduction to Sequentially Markovian Coalescent Methods for Estimating Demographic History From Genomic Data.” Ecology and Evolution, vol. 10, no. 1, Dec. 2019, pp. 579–89, doi:10.1002/ece3.5888.
Unm-Carc. “QuickBytes/psmc_quickbyte.md at Master · UNM-CARC/QuickBytes.” GitHub, github.com/UNM-CARC/QuickBytes/blob/master/psmc_quickbyte.md.
sessionInfo()R version 4.4.2 (2024-10-31)
Platform: aarch64-apple-darwin20
Running under: macOS Sequoia 15.6.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Asia/Tokyo
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
loaded via a namespace (and not attached):
[1] vctrs_0.6.5 cli_3.6.5 knitr_1.50 rlang_1.1.6
[5] xfun_0.52 stringi_1.8.7 promises_1.3.3 jsonlite_2.0.0
[9] workflowr_1.7.1 glue_1.8.0 rprojroot_2.0.4 git2r_0.36.2
[13] htmltools_0.5.8.1 httpuv_1.6.16 sass_0.4.10 rmarkdown_2.29
[17] tibble_3.3.0 evaluate_1.0.4 jquerylib_0.1.4 fastmap_1.2.0
[21] yaml_2.3.10 lifecycle_1.0.4 whisker_0.4.1 stringr_1.5.1
[25] compiler_4.4.2 fs_1.6.6 pkgconfig_2.0.3 Rcpp_1.0.14
[29] rstudioapi_0.17.1 later_1.4.2 digest_0.6.37 R6_2.6.1
[33] pillar_1.10.2 magrittr_2.0.3 bslib_0.9.0 tools_4.4.2
[37] cachem_1.1.0