Enrichment Analysis: Mutant Vs Wild-Type
Steve Pederson
07 March, 2020
Last updated: 2020-03-07
Checks: 7 0
Knit directory: 20170327_Psen2S4Ter_RNASeq/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200119) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Untracked files:
Untracked: output/AllResults.xlsx
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 65ae03b | Steve Ped | 2020-03-07 | Typo |
| Rmd | 3b120d9 | Steve Ped | 2020-03-07 | Corrected table justifications |
| html | a5cdf5d | Steve Ped | 2020-03-06 | Finished Revisions |
| Rmd | 2772d42 | Steve Ped | 2020-03-06 | Corrected UpSet names |
| Rmd | a9781d9 | Steve Ped | 2020-03-06 | Fixed duplicate labels |
| Rmd | 154085b | Steve Ped | 2020-03-06 | Reduced ranked lists to fry only |
| Rmd | 2aa6e06 | Steve Ped | 2020-03-06 | Removed gsea, camera & combining data. Now only using fry + goseq |
| Rmd | 433a729 | Steve Ped | 2020-03-06 | Prepared to remove EGSEA-type approach |
| Rmd | 956c065 | Steve Ped | 2020-03-05 | Revised GO exploration |
| html | 1385ceb | Steve Ped | 2020-03-05 | Compiled after renaming columns |
| Rmd | 2c56cef | Steve Ped | 2020-03-05 | Tidied column names for enrichment output |
| html | 7fe1505 | Steve Ped | 2020-03-05 | Updated all analyses & heatmaps after changing to cpmPostNorm instead of fit\(fitted.values for enrichment/heatmaps</td> </tr> <tr> <td>Rmd</td> <td><a href="https://github.com/UofABioinformaticsHub/20170327_Psen2S4Ter_RNASeq/blob/bad81c1731ba8bc4487d25ec2719e92ae6917e4a/analysis/3_Enrichment_MutantVsWT.Rmd" target="_blank">bad81c1</a></td> <td>Steve Ped</td> <td>2020-03-05</td> <td>Updated MutVsWt UpSet plots for KEGG/HALLMARK</td> </tr> <tr> <td>Rmd</td> <td><a href="https://github.com/UofABioinformaticsHub/20170327_Psen2S4Ter_RNASeq/blob/377410a12ca13a7e233e39fb2f77f165e846d830/analysis/3_Enrichment_MutantVsWT.Rmd" target="_blank">377410a</a></td> <td>Steve Ped</td> <td>2020-03-05</td> <td>Finished revision of Enrichment Analysis</td> </tr> <tr> <td>Rmd</td> <td><a href="https://github.com/UofABioinformaticsHub/20170327_Psen2S4Ter_RNASeq/blob/c0b89ed8f761b0c7222538c353d21cd5aa402b94/analysis/3_Enrichment_MutantVsWT.Rmd" target="_blank">c0b89ed</a></td> <td>Steve Ped</td> <td>2020-03-05</td> <td>Reran after changing to cpmPotNorm instead of the incorrect fit\)fitted.values |
| html | 7e59d3c | Steve Ped | 2020-02-20 | Recompiled |
| Rmd | 4ede752 | Steve Ped | 2020-02-20 | Added headings |
| html | 0168824 | Steve Ped | 2020-02-20 | Rebult after resizing |
| Rmd | de9c4b3 | Steve Ped | 2020-02-20 | Resized heatmaps again |
| html | 86ffa07 | Steve Ped | 2020-02-20 | Updated saample layout figure |
| Rmd | 4d2af23 | Steve Ped | 2020-02-20 | Modified heatmap sizes & initial network layout |
| Rmd | 8fe35b0 | Steve Ped | 2020-02-20 | Added multiple heatmaps & UpSet plots |
| html | 679b10d | Steve Ped | 2020-02-19 | Rendered OXPHOS Plot |
| Rmd | 66ba4e0 | Steve Ped | 2020-02-19 | Added OXPHOS plot |
| html | e0288c6 | Steve Ped | 2020-02-19 | Generated enrichment tables |
| Rmd | 541fbf0 | Steve Ped | 2020-02-19 | Revised Hom Vs Het Enrichment |
| Rmd | 77f167d | Steve Ped | 2020-02-19 | Revised enrichment |
| html | 876e40f | Steve Ped | 2020-02-17 | Compiled after minor corrections |
| Rmd | 53ed0e3 | Steve Ped | 2020-02-17 | Corrected Ensembl Release |
| Rmd | f55be85 | Steve Ped | 2020-02-17 | Added commas |
| Rmd | 8f29458 | Steve Ped | 2020-02-17 | Minor tweaks to formatting |
| html | 9104ecd | Steve Ped | 2020-01-28 | First draft of Hom Vs Het |
| Rmd | 207cdc8 | Steve Ped | 2020-01-28 | Added code for Hom Vs Het Enrichment |
| Rmd | 468e6e3 | Steve Ped | 2020-01-28 | Ran enrichment of Mut Vs WT |
| html | 468e6e3 | Steve Ped | 2020-01-28 | Ran enrichment of Mut Vs WT |
| Rmd | 3a9933c | Steve Ped | 2020-01-28 | Finished Enrichment analysis on MutVsWt |
| html | 3a9933c | Steve Ped | 2020-01-28 | Finished Enrichment analysis on MutVsWt |
Setup
library(tidyverse)
library(magrittr)
library(edgeR)
library(scales)
library(pander)
library(goseq)
library(msigdbr)
library(AnnotationDbi)
library(RColorBrewer)
library(ngsReports)
library(UpSetR)
library(pheatmap)theme_set(theme_bw())
panderOptions("table.split.table", Inf)
panderOptions("table.style", "rmarkdown")
panderOptions("big.mark", ",")samples <- read_csv("data/samples.csv") %>%
distinct(sampleName, .keep_all = TRUE) %>%
dplyr::select(sample = sampleName, sampleID, genotype) %>%
mutate(
genotype = factor(genotype, levels = c("WT", "Het", "Hom")),
mutant = genotype %in% c("Het", "Hom"),
homozygous = genotype == "Hom"
)
genoCols <- samples$genotype %>%
levels() %>%
length() %>%
brewer.pal("Set1") %>%
setNames(levels(samples$genotype))dgeList <- here::here("data/dgeList.rds") %>% read_rds()
cpmPostNorm <- here::here("data/cpmPostNorm.rds") %>% read_rds()
entrezGenes <- dgeList$genes %>%
dplyr::filter(!is.na(entrezid)) %>%
unnest(entrezid) %>%
dplyr::rename(entrez_gene = entrezid)deTable <- here::here("output", "psen2VsWT.csv") %>%
read_csv() %>%
mutate(
entrezid = dgeList$genes$entrezid[gene_id]
)formatP <- function(p, m = 0.0001){
out <- rep("", length(p))
out[p < m] <- sprintf("%.2e", p[p<m])
out[p >= m] <- sprintf("%.4f", p[p>=m])
out
}Introduction
Enrichment analysis for this dataset present some challenges. Despite normalisation to account for gene length and GC bias, some appeared to still be present in the final results. In addition, the confounding of incomplete rRNA removal with genotype may lead to other distortions in both DE genes and ranking statistics.
Two steps for enrichment analysis will be undertaken.
- Testing for enrichment within discrete sets of DE genes as defined in the previous steps
- Testing for enrichment using the complete set of results using
fry.
Testing for enrichment within discrete gene sets will be performed using goseq as this allows for the incorporation of a single covariate as a predictor of differential expression. GC content, gene length and correlation with rRNA removal can all be supplied as separate covariates.
For enrichment within larger gene lists, fry can take into account inter-gene correlations. Values supplied will be logCPM for each gene/sample after being adjusted for GC and length biases.
Databases used for testing
Data was sourced using the msigdbr package. The initial database used for testing was the Hallmark Gene Sets, with mappings from gene-set to EntrezGene IDs performed by the package authors.
Hallmark Gene Sets
hm <- msigdbr("Danio rerio", category = "H") %>%
left_join(entrezGenes) %>%
dplyr::filter(!is.na(gene_id)) %>%
distinct(gs_name, gene_id, .keep_all = TRUE)
hmByGene <- hm %>%
split(f = .$gene_id) %>%
lapply(extract2, "gs_name")
hmByID <- hm %>%
split(f = .$gs_name) %>%
lapply(extract2, "gene_id")Mappings are required from gene to pathway, and Ensembl identifiers were used to map from gene to pathway, based on the mappings in the previously used annotations (Ensembl Release 98). A total of 3,459 Ensembl IDs were mapped to pathways from the hallmark gene sets.
KEGG Gene Sets
kg <- msigdbr("Danio rerio", category = "C2", subcategory = "CP:KEGG") %>%
left_join(entrezGenes) %>%
dplyr::filter(!is.na(gene_id)) %>%
distinct(gs_name, gene_id, .keep_all = TRUE)
kgByGene <- kg %>%
split(f = .$gene_id) %>%
lapply(extract2, "gs_name")
kgByID <- kg %>%
split(f = .$gs_name) %>%
lapply(extract2, "gene_id")The same mapping process was applied to KEGG gene sets. A total of 3,614 Ensembl IDs were mapped to pathways from the KEGG gene sets.
Gene Ontology Gene Sets
goSummaries <- url("https://uofabioinformaticshub.github.io/summaries2GO/data/goSummaries.RDS") %>%
readRDS() %>%
mutate(
Term = Term(id),
gs_name = Term %>% str_to_upper() %>% str_replace_all("[ -]", "_"),
gs_name = paste0("GO_", gs_name)
)
minPath <- 3go <- msigdbr("Danio rerio", category = "C5") %>%
left_join(entrezGenes) %>%
dplyr::filter(!is.na(gene_id)) %>%
left_join(goSummaries) %>%
dplyr::filter(shortest_path >= minPath) %>%
distinct(gs_name, gene_id, .keep_all = TRUE)
goByGene <- go %>%
split(f = .$gene_id) %>%
lapply(extract2, "gs_name")
goByID <- go %>%
split(f = .$gs_name) %>%
lapply(extract2, "gene_id")For analysis of gene-sets from the GO database, gene-sets were restricted to those with 3 or more steps back to the ontology root terms. A total of 11,245 Ensembl IDs were mapped to pathways from restricted database of 8,834 GO gene sets.
gsSizes <- bind_rows(hm, kg, go) %>%
dplyr::select(gs_name, gene_symbol, gene_id) %>%
chop(c(gene_symbol, gene_id)) %>%
mutate(
gs_size = vapply(gene_symbol, length, integer(1)),
de_id = lapply(
X = gene_id,
FUN = intersect,
y = dplyr::filter(deTable, DE)$gene_id
),
de_size = vapply(de_id, length, integer(1))
)Enrichment in the DE Gene Set
The first step of analysis using goseq, regardless of the gene-set, is estimation of the probability weight function (PWF) which quantifies the probability of a gene being considered as DE based on a single covariate. As GC content and length should have been accounted for during conditional-quantile normalisation, these were not required for any bias. However, the gene-level correlations with rRNA contamination were instead used a predictor of bias in selection of a gene as being DE.
rawFqc <- list.files(
path = here::here("data/0_rawData/FastQC/"),
pattern = "zip",
full.names = TRUE
) %>%
FastqcDataList()
gc <- getModule(rawFqc, "Per_sequence_GC") %>%
group_by(Filename) %>%
mutate(Freq = Count / sum(Count)) %>%
ungroup()
gcDev <- gc %>%
left_join(getGC(gcTheoretical, "Drerio", "Trans")) %>%
mutate(
sample = str_remove(Filename, "_R[12].fastq.gz"),
resid = Freq - Drerio
) %>%
left_join(samples) %>%
group_by(sample) %>%
summarise(
ss = sum(resid^2),
n = n(),
sd = sqrt(ss / (n - 1))
)riboVec <- structure(gcDev$sd, names = gcDev$sample)
riboCors <- cpm(dgeList, log = TRUE) %>%
apply(1, function(x){
cor(x, riboVec[names(x)])
})Values were calculated as per the previous steps, using the logCPM values for each gene, with the sample-level standard deviations from the theoretical GC distribution being used as a measure of rRNA contamination. For estimation of the probability weight function, squared correlations were used to place negative and positive correlations on the same scale. This accounts for genes which are both negatively & positively biased by the presence of excessive rRNA. Clearly, the confounding of genotype with rRNA means that some genes driven by the genuine biology may be down-weighted under this approach.
riboPwf <- deTable %>%
mutate(riboCors = riboCors[gene_id]^2) %>%
dplyr::select(gene_id, DE, riboCors) %>%
distinct(gene_id, .keep_all = TRUE) %>%
with(
nullp(
DEgenes = structure(
as.integer(DE), names = gene_id
),
genome = "danRer10",
id = "ensGene",
bias.data = riboCors,
plot.fit = FALSE
)
)
plotPWF(riboPwf, main = "Bias from rRNA proportions")
Using this approach, it was clear that correlation with rRNA proportions significantly biased the probability of a gene being considered as DE.
| Version | Author | Date |
|---|---|---|
| 468e6e3 | Steve Ped | 2020-01-28 |
All gene-sets were then tested using this PWF.
Hallmark Gene Sets
hmRiboGoseq <- goseq(riboPwf, gene2cat = hmByGene) %>%
as_tibble %>%
dplyr::filter(numDEInCat > 0) %>%
mutate(
adjP = p.adjust(over_represented_pvalue, method = "bonf"),
FDR = p.adjust(over_represented_pvalue, method = "fdr")
) %>%
dplyr::select(-contains("under")) %>%
dplyr::rename(
gs_name = category,
PValue = over_represented_pvalue
)No gene-sets achieved significance in the DE genes with the lowest FDR being 41%
KEGG Gene Sets
kgRiboGoseq <- goseq(riboPwf, gene2cat = kgByGene) %>%
as_tibble %>%
dplyr::filter(numDEInCat > 0) %>%
mutate(
adjP = p.adjust(over_represented_pvalue, method = "bonf"),
FDR = p.adjust(over_represented_pvalue, method = "fdr")
) %>%
dplyr::select(-contains("under")) %>%
dplyr::rename(
gs_name = category,
PValue = over_represented_pvalue
)kgRiboGoseq %>%
dplyr::slice(1:5) %>%
mutate(
p = formatP(PValue),
adjP = formatP(adjP),
FDR = formatP(FDR)
) %>%
dplyr::select(
`Gene Set` = gs_name,
DE = numDEInCat,
`Set Size` = numInCat,
PValue,
`p~bonf~` = adjP,
`p~FDR~` = FDR
) %>%
pander(
justify = "lrrrrr",
caption = paste(
"The", nrow(.), "most highly-ranked KEGG pathways.",
"Bonferroni-adjusted p-values are the most stringent and give high",
"confidence when below 0.05."
)
)| Gene Set | DE | Set Size | PValue | pbonf | pFDR |
|---|---|---|---|---|---|
| KEGG_RIBOSOME | 26 | 80 | 4.754e-09 | 5.32e-07 | 5.32e-07 |
| KEGG_PRIMARY_IMMUNODEFICIENCY | 2 | 15 | 0.01642 | 1.0000 | 0.7173 |
| KEGG_CYSTEINE_AND_METHIONINE_METABOLISM | 4 | 26 | 0.02959 | 1.0000 | 0.7173 |
| KEGG_ASTHMA | 1 | 3 | 0.04088 | 1.0000 | 0.7173 |
| KEGG_RETINOL_METABOLISM | 3 | 25 | 0.0512 | 1.0000 | 0.7173 |
Notably, the KEGG gene-set for Ribosomal genes was detected as enriched in the set of DE genes, with no other KEGG gene-sets being considered significant.
GO Gene Sets
goRiboGoseq <- goseq(riboPwf, gene2cat = goByGene) %>%
as_tibble %>%
dplyr::filter(numDEInCat > 0) %>%
mutate(
adjP = p.adjust(over_represented_pvalue, method = "bonf"),
FDR = p.adjust(over_represented_pvalue, method = "fdr")
) %>%
dplyr::select(-contains("under")) %>%
dplyr::rename(
gs_name = category,
PValue = over_represented_pvalue
)goRiboGoseq %>%
dplyr::filter(adjP < 0.05) %>%
mutate(
p = formatP(PValue),
adjP = formatP(adjP),
FDR = formatP(FDR)
) %>%
dplyr::select(
`Gene Set` = gs_name,
DE = numDEInCat,
`Set Size` = numInCat,
PValue,
`p~bonf~` = adjP,
`p~FDR~` = FDR
) %>%
pander(
justify = "lrrrrr",
caption = paste(
"*The", nrow(.), "most highly-ranked GO terms.",
"Bonferroni-adjusted p-values are the most stringent and give high",
"confidence when below 0.05, with all terms here reaching this threshold.",
"However, most terms once again indicate the presence of rRNA.*"
)
)| Gene Set | DE | Set Size | PValue | pbonf | pFDR |
|---|---|---|---|---|---|
| GO_COTRANSLATIONAL_PROTEIN_TARGETING_TO_MEMBRANE | 28 | 93 | 7.36e-10 | 2.83e-06 | 2.03e-06 |
| GO_CYTOSOLIC_RIBOSOME | 27 | 97 | 1.056e-09 | 4.06e-06 | 2.03e-06 |
| GO_ESTABLISHMENT_OF_PROTEIN_LOCALIZATION_TO_ENDOPLASMIC_RETICULUM | 27 | 105 | 1.6e-08 | 6.16e-05 | 2.05e-05 |
| GO_TRANSLATIONAL_INITIATION | 29 | 177 | 2.178e-07 | 0.0008 | 0.0002 |
| GO_PROTEIN_TARGETING_TO_MEMBRANE | 30 | 171 | 2.533e-07 | 0.0010 | 0.0002 |
| GO_PROTEIN_LOCALIZATION_TO_ENDOPLASMIC_RETICULUM | 27 | 128 | 3.849e-07 | 0.0015 | 0.0002 |
| GO_CYTOSOLIC_PART | 30 | 206 | 4.424e-07 | 0.0017 | 0.0002 |
| GO_VIRAL_GENE_EXPRESSION | 29 | 172 | 6.939e-07 | 0.0027 | 0.0003 |
| GO_NUCLEAR_TRANSCRIBED_MRNA_CATABOLIC_PROCESS | 30 | 183 | 7.911e-07 | 0.0030 | 0.0003 |
| GO_RIBOSOMAL_SUBUNIT | 28 | 175 | 1.99e-06 | 0.0077 | 0.0008 |
| GO_RIBOSOME | 30 | 210 | 2.659e-06 | 0.0102 | 0.0009 |
| GO_CYTOSOLIC_LARGE_RIBOSOMAL_SUBUNIT | 15 | 51 | 2.826e-06 | 0.0109 | 0.0009 |
| GO_SYMPORTER_ACTIVITY | 10 | 94 | 7.571e-06 | 0.0291 | 0.0022 |
| GO_ANION_TRANSMEMBRANE_TRANSPORTER_ACTIVITY | 17 | 228 | 1.23e-05 | 0.0473 | 0.0034 |
Enrichment Testing on the Complete Set of Genes
Hallmark Gene Sets
hmFry <- cpmPostNorm %>%
fry(
index = hmByID,
design = dgeList$design,
contrast = "mutant",
sort = "directional"
) %>%
rownames_to_column("gs_name") %>%
as_tibble()hmFry %>%
dplyr::filter(
PValue < 0.01
) %>%
left_join(gsSizes) %>%
mutate(gs_name = str_remove(gs_name, "HALLMARK_")) %>%
dplyr::select(
`Hallmark Gene Set` = gs_name,
`Expressed Genes` = NGenes,
`DE Genes` = de_size,
Direction,
PValue, FDR
) %>%
pander(
justify = "lrrlrr",
caption = "Enriched Hallmark Gene Sets, using a threshold of p < 0.01, which corresponds to an FDR of 0.02"
)| Hallmark Gene Set | Expressed Genes | DE Genes | Direction | PValue | FDR |
|---|---|---|---|---|---|
| UV_RESPONSE_UP | 141 | 7 | Up | 4.187e-06 | 0.0002094 |
| XENOBIOTIC_METABOLISM | 146 | 11 | Up | 8.577e-05 | 0.001756 |
| REACTIVE_OXYGEN_SPECIES_PATHWAY | 45 | 5 | Up | 0.0001128 | 0.001756 |
| FATTY_ACID_METABOLISM | 135 | 6 | Up | 0.0001574 | 0.001756 |
| MTORC1_SIGNALING | 194 | 11 | Up | 0.0001756 | 0.001756 |
| GLYCOLYSIS | 172 | 8 | Up | 0.0002864 | 0.002387 |
| PEROXISOME | 95 | 2 | Up | 0.0004238 | 0.003027 |
| ADIPOGENESIS | 186 | 13 | Up | 0.0005475 | 0.003236 |
| BILE_ACID_METABOLISM | 86 | 1 | Up | 0.0006345 | 0.003236 |
| PROTEIN_SECRETION | 92 | 3 | Up | 0.0006473 | 0.003236 |
| COAGULATION | 80 | 3 | Up | 0.0008365 | 0.003624 |
| UNFOLDED_PROTEIN_RESPONSE | 113 | 4 | Up | 0.0008697 | 0.003624 |
| CHOLESTEROL_HOMEOSTASIS | 69 | 2 | Up | 0.001557 | 0.005666 |
| NOTCH_SIGNALING | 29 | 0 | Up | 0.001623 | 0.005666 |
| ESTROGEN_RESPONSE_LATE | 153 | 3 | Up | 0.0017 | 0.005666 |
| MYC_TARGETS_V1 | 192 | 15 | Up | 0.0021 | 0.005993 |
| HYPOXIA | 169 | 9 | Up | 0.002131 | 0.005993 |
| OXIDATIVE_PHOSPHORYLATION | 201 | 15 | Up | 0.002158 | 0.005993 |
| EPITHELIAL_MESENCHYMAL_TRANSITION | 156 | 7 | Up | 0.003453 | 0.009086 |
| TNFA_SIGNALING_VIA_NFKB | 149 | 7 | Up | 0.004565 | 0.01141 |
| IL2_STAT5_SIGNALING | 147 | 2 | Up | 0.005095 | 0.01213 |
| ANDROGEN_RESPONSE | 90 | 2 | Up | 0.006364 | 0.01446 |
| PI3K_AKT_MTOR_SIGNALING | 98 | 2 | Up | 0.00925 | 0.02011 |
Exploration of significant Hallmark Gene Sets
hmFry %>%
left_join(gsSizes) %>%
dplyr::filter(de_size >= 5, FDR < 0.05) %>%
dplyr::select(gs_name, de_id) %>%
unnest(de_id) %>%
mutate(
gs_name = str_remove(gs_name, "HALLMARK_"),
gs_name = fct_lump(gs_name, n = 14)
) %>%
split(f = .$gs_name) %>%
lapply(magrittr::extract2, "de_id") %>%
fromList() %>%
upset(
nsets = length(.),
nintersects = 20,
order.by = "freq",
mb.ratio = c(0.6, 0.4),
sets.x.label = "Number Of DE Genes"
)
UpSet plot indicating distribution of DE genes within all significant HALLMARK gene sets. Gene sets were restricted to those with an FDR < 0.05 and at least 5 DE genes. The plot is truncated at the right for simplicity. Most gene sets seem relatively independent of each other with regard to DE genes.
| Version | Author | Date |
|---|---|---|
| a5cdf5d | Steve Ped | 2020-03-06 |
hmHeat <- hmFry %>%
left_join(gsSizes) %>%
dplyr::filter(de_size >= 12, FDR < 0.05) %>%
dplyr::select(gs_name, de_id) %>%
unnest(de_id) %>%
left_join(dgeList$genes, by = c("de_id" = "gene_id")) %>%
dplyr::select(gs_name, de_id, gene_name) %>%
mutate(belongs = TRUE) %>%
pivot_wider(
id_cols = c(de_id, gene_name),
names_from = gs_name,
values_from = belongs,
values_fill = list(belongs = FALSE)
) %>%
left_join(
cpmPostNorm %>%
as.data.frame() %>%
rownames_to_column("de_id")
)
hmHeat %>%
dplyr::select(gene_name, starts_with("Ps")) %>%
as.data.frame() %>%
column_to_rownames("gene_name") %>%
as.matrix() %>%
pheatmap(
color = viridis_pal(option = "magma")(100),
legend_breaks = c(seq(-2, 8, by = 2), max(.)),
legend_labels = c(seq(-2, 8, by = 2), "logCPM\n"),
labels_col = hmHeat %>%
dplyr::select(starts_with("Ps")) %>%
colnames() %>%
enframe(name = NULL) %>%
dplyr::rename(sample = value) %>%
left_join(dgeList$samples) %>%
dplyr::select(sample, sampleID) %>%
with(
structure(sampleID, names = sample)
),
cutree_rows = 6,
cutree_cols = 2,
annotation_names_row = FALSE,
annotation_row = hmHeat %>%
dplyr::select(gene_name, starts_with("HALLMARK_")) %>%
mutate_at(vars(starts_with("HALL")), as.character) %>%
as.data.frame() %>%
column_to_rownames("gene_name") %>%
set_colnames(
str_remove(colnames(.), "HALLMARK_")
)
)
Gene expression patterns for all DE genes in HALLMARK gene sets, containing more than 12 DE genes.
KEGG Gene Sets
kgFry <- cpmPostNorm %>%
fry(
index = kgByID,
design = dgeList$design,
contrast = "mutant",
sort = "directional"
) %>%
rownames_to_column("gs_name") %>%
as_tibble()kgFry %>%
dplyr::filter(
PValue < 0.005
) %>%
left_join(gsSizes) %>%
mutate(gs_name = str_remove(gs_name, "HALLMARK_")) %>%
dplyr::select(
`KEGG Gene Set` = gs_name,
`Expressed Genes` = NGenes,
`DE Genes` = de_size,
Direction,
PValue, FDR
) %>%
pander(
justify = "lrrlrr",
caption = "Enriched KEGG Gene Sets, using a threshold p < 0.005, which corresponds to an FDR of 0.02"
)| KEGG Gene Set | Expressed Genes | DE Genes | Direction | PValue | FDR |
|---|---|---|---|---|---|
| KEGG_GLUTATHIONE_METABOLISM | 36 | 4 | Up | 1.313e-05 | 0.002442 |
| KEGG_GLYCINE_SERINE_AND_THREONINE_METABOLISM | 24 | 2 | Up | 9.541e-05 | 0.005307 |
| KEGG_ARGININE_AND_PROLINE_METABOLISM | 39 | 2 | Up | 9.545e-05 | 0.005307 |
| KEGG_LIMONENE_AND_PINENE_DEGRADATION | 8 | 0 | Up | 0.0001542 | 0.005307 |
| KEGG_PRIMARY_IMMUNODEFICIENCY | 15 | 2 | Down | 0.0001638 | 0.005307 |
| KEGG_PORPHYRIN_AND_CHLOROPHYLL_METABOLISM | 25 | 0 | Up | 0.0002012 | 0.005307 |
| KEGG_ASCORBATE_AND_ALDARATE_METABOLISM | 7 | 0 | Up | 0.000218 | 0.005307 |
| KEGG_BETA_ALANINE_METABOLISM | 18 | 1 | Up | 0.0002525 | 0.005307 |
| KEGG_GLYCEROLIPID_METABOLISM | 37 | 1 | Up | 0.0002568 | 0.005307 |
| KEGG_PPAR_SIGNALING_PATHWAY | 53 | 1 | Up | 0.0002947 | 0.005397 |
| KEGG_FATTY_ACID_METABOLISM | 38 | 1 | Up | 0.0003304 | 0.005397 |
| KEGG_BUTANOATE_METABOLISM | 26 | 0 | Up | 0.0003879 | 0.005397 |
| KEGG_T_CELL_RECEPTOR_SIGNALING_PATHWAY | 79 | 3 | Down | 0.0004045 | 0.005397 |
| KEGG_AMINOACYL_TRNA_BIOSYNTHESIS | 37 | 0 | Up | 0.0004062 | 0.005397 |
| KEGG_TYROSINE_METABOLISM | 29 | 2 | Up | 0.0004776 | 0.005698 |
| KEGG_METABOLISM_OF_XENOBIOTICS_BY_CYTOCHROME_P450 | 26 | 3 | Up | 0.0004901 | 0.005698 |
| KEGG_DRUG_METABOLISM_CYTOCHROME_P450 | 26 | 3 | Up | 0.0005606 | 0.005827 |
| KEGG_PYRUVATE_METABOLISM | 27 | 1 | Up | 0.0006084 | 0.005827 |
| KEGG_CYSTEINE_AND_METHIONINE_METABOLISM | 26 | 4 | Up | 0.0006305 | 0.005827 |
| KEGG_BIOSYNTHESIS_OF_UNSATURATED_FATTY_ACIDS | 21 | 0 | Up | 0.0006464 | 0.005827 |
| KEGG_GLYOXYLATE_AND_DICARBOXYLATE_METABOLISM | 13 | 1 | Up | 0.0006579 | 0.005827 |
| KEGG_SELENOAMINO_ACID_METABOLISM | 22 | 0 | Up | 0.0007171 | 0.005874 |
| KEGG_EPITHELIAL_CELL_SIGNALING_IN_HELICOBACTER_PYLORI_INFECTION | 60 | 0 | Up | 0.0007358 | 0.005874 |
| KEGG_VIBRIO_CHOLERAE_INFECTION | 47 | 1 | Up | 0.000758 | 0.005874 |
| KEGG_LYSINE_DEGRADATION | 39 | 0 | Up | 0.0009213 | 0.006855 |
| KEGG_PHENYLALANINE_METABOLISM | 11 | 1 | Up | 0.0009648 | 0.006866 |
| KEGG_RENIN_ANGIOTENSIN_SYSTEM | 8 | 1 | Up | 0.001033 | 0.006866 |
| KEGG_NITROGEN_METABOLISM | 13 | 1 | Up | 0.001034 | 0.006866 |
| KEGG_COMPLEMENT_AND_COAGULATION_CASCADES | 27 | 1 | Up | 0.00112 | 0.007181 |
| KEGG_RIBOSOME | 80 | 26 | Up | 0.001277 | 0.007917 |
| KEGG_PEROXISOME | 70 | 0 | Up | 0.00135 | 0.008102 |
| KEGG_VALINE_LEUCINE_AND_ISOLEUCINE_BIOSYNTHESIS | 10 | 0 | Up | 0.00144 | 0.008369 |
| KEGG_PATHOGENIC_ESCHERICHIA_COLI_INFECTION | 44 | 3 | Up | 0.001755 | 0.009894 |
| KEGG_VALINE_LEUCINE_AND_ISOLEUCINE_DEGRADATION | 40 | 0 | Up | 0.001835 | 0.01004 |
| KEGG_RETINOL_METABOLISM | 25 | 3 | Up | 0.001902 | 0.01011 |
| KEGG_PURINE_METABOLISM | 135 | 4 | Up | 0.001958 | 0.01011 |
| KEGG_LEUKOCYTE_TRANSENDOTHELIAL_MIGRATION | 87 | 1 | Up | 0.002044 | 0.01018 |
| KEGG_ALZHEIMERS_DISEASE | 141 | 12 | Up | 0.002128 | 0.01018 |
| KEGG_CITRATE_CYCLE_TCA_CYCLE | 26 | 2 | Up | 0.002141 | 0.01018 |
| KEGG_N_GLYCAN_BIOSYNTHESIS | 45 | 1 | Up | 0.00219 | 0.01018 |
| KEGG_OXIDATIVE_PHOSPHORYLATION | 119 | 14 | Up | 0.002373 | 0.01065 |
| KEGG_PROPANOATE_METABOLISM | 28 | 1 | Up | 0.002406 | 0.01065 |
| KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION | 15 | 0 | Up | 0.002581 | 0.01116 |
| KEGG_PROTEASOME | 42 | 2 | Up | 0.00284 | 0.01187 |
| KEGG_GLYCOLYSIS_GLUCONEOGENESIS | 39 | 3 | Up | 0.002871 | 0.01187 |
| KEGG_B_CELL_RECEPTOR_SIGNALING_PATHWAY | 64 | 3 | Down | 0.003085 | 0.01247 |
| KEGG_GLYCEROPHOSPHOLIPID_METABOLISM | 56 | 1 | Up | 0.004306 | 0.01704 |
| KEGG_ABC_TRANSPORTERS | 32 | 0 | Up | 0.004993 | 0.01935 |
Exploration of Significant KEGG Gene Sets
kgFry %>%
left_join(gsSizes) %>%
dplyr::filter(de_size >= 4, FDR < 0.05) %>%
dplyr::select(gs_name, de_id) %>%
unnest(de_id) %>%
mutate(
gs_name = str_remove(gs_name, "KEGG_"),
gs_name = fct_lump(gs_name, n = 14)
) %>%
split(f = .$gs_name) %>%
lapply(magrittr::extract2, "de_id") %>%
fromList() %>%
upset(
nsets = length(.),
nintersects = 20,
order.by = "freq",
mb.ratio = c(0.6, 0.4),
sets.x.label = "Number Of DE Genes"
)
UpSet plot indicating distribution of DE genes within all significant terms from the KEGG gene sets. Gene sets were restricted to those with an FDR < 0.05 and four or more DE genes. There is considerable overlap between DE genes in Oxidative Phosphorylation and both Parkinson’s and Huntington’s Disease, indicating that these terms essentially capture the same expression signal within this dataset.
| Version | Author | Date |
|---|---|---|
| a5cdf5d | Steve Ped | 2020-03-06 |
riboOx <- kg %>%
dplyr::filter(
grepl("OXIDATIVE_PHOS", gs_name) | grepl("RIBOSOME", gs_name),
gene_id %in% dplyr::filter(deTable, DE)$gene_id
) %>%
mutate(gs_name = str_remove(gs_name, "(KEGG|HALLMARK)_")) %>%
distinct(gs_name, gene_id) %>%
left_join(
cpmPostNorm %>%
as.data.frame() %>%
rownames_to_column("gene_id") %>%
as_tibble()
) %>%
pivot_longer(cols = starts_with("Ps"), names_to = "sample", values_to = "CPM") %>%
left_join(dgeList$samples) %>%
left_join(dgeList$genes) %>%
dplyr::select(gs_name, gene_name, sampleID, genotype, CPM) %>%
pivot_wider(
id_cols = c(gs_name, gene_name),
values_from = CPM,
names_from = sampleID
)
riboOx %>%
dplyr::select(-gs_name) %>%
as.data.frame() %>%
column_to_rownames("gene_name") %>%
as.matrix() %>%
pheatmap(
annotation_row = data.frame(
GeneSet = riboOx$gs_name %>% str_replace("OXI.+", "OXPHOS"),
row.names = riboOx$gene_name
),
color = viridis_pal(option = "magma")(100),
legend_breaks = c(seq(-2, 8, by = 2), max(.)),
legend_labels = c(seq(-2, 8, by = 2), "logCPM\n"),
annotation_names_row = FALSE,
cutree_rows = 3,
cutree_cols = 2
)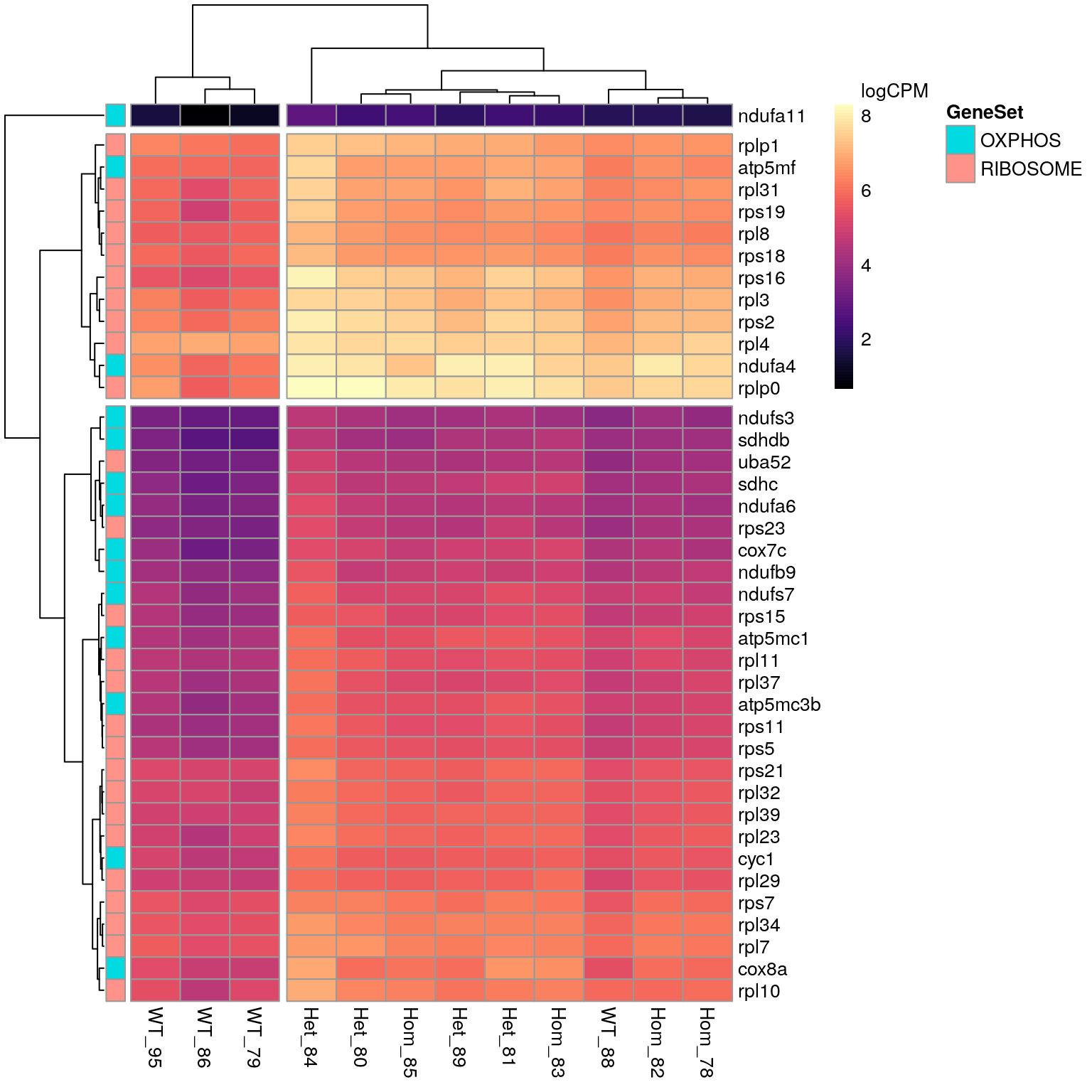
Given that the largest gene sets within the KEGG results were Oxidative Phosphorylation and Ribosomal gene sets, all DE genes associated with these KEGG terms are displayed. All genes appear to show increased expression in mutant samples.
GO Gene Sets
goFry <- cpmPostNorm %>%
fry(
index = goByID,
design = dgeList$design,
contrast = "mutant",
sort = "directional"
) %>%
rownames_to_column("gs_name") %>%
as_tibble()goFry %>%
left_join(gsSizes) %>%
left_join(distinct(go, gs_name, shortest_path)) %>%
dplyr::filter(
FDR < 0.01,
shortest_path > 3,
NGenes < 1000,
de_size >= 5
) %>%
dplyr::select(
`GO Gene Set` = gs_name,
`Expressed Genes` = NGenes,
`DE Genes` = de_size,
Direction,
PValue, FDR
) %>%
pander(
justify = "lrrlrr",
caption = "Enriched GO Gene Sets, using the thresholds FDR < 0.01, at least 5 DE genes, a set size < 1000 and more than 3 steps back to the ontology root."
)| GO Gene Set | Expressed Genes | DE Genes | Direction | PValue | FDR |
|---|---|---|---|---|---|
| GO_PEPTIDYL_LYSINE_TRIMETHYLATION | 42 | 5 | Down | 6.682e-06 | 0.001904 |
| GO_FATTY_ACID_BIOSYNTHETIC_PROCESS | 96 | 6 | Up | 9.201e-06 | 0.002258 |
| GO_ORGANIC_ACID_TRANSPORT | 240 | 14 | Up | 3.617e-05 | 0.003991 |
| GO_ORGANIC_ACID_BIOSYNTHETIC_PROCESS | 198 | 12 | Up | 4.586e-05 | 0.004057 |
| GO_PEPTIDYL_LYSINE_METHYLATION | 111 | 5 | Down | 4.848e-05 | 0.004057 |
| GO_RESPONSE_TO_CORTICOSTEROID | 121 | 5 | Up | 4.854e-05 | 0.004057 |
| GO_PEPTIDYL_LYSINE_ACETYLATION | 144 | 5 | Down | 6.245e-05 | 0.004211 |
| GO_MONOCARBOXYLIC_ACID_BIOSYNTHETIC_PROCESS | 127 | 7 | Up | 7.071e-05 | 0.004534 |
| GO_REGULATION_OF_LIPID_BIOSYNTHETIC_PROCESS | 138 | 6 | Up | 7.186e-05 | 0.004534 |
| GO_RESPONSE_TO_ALCOHOL | 177 | 7 | Up | 7.293e-05 | 0.004534 |
| GO_HISTONE_METHYLATION | 118 | 5 | Down | 7.92e-05 | 0.004536 |
| GO_MONOCARBOXYLIC_ACID_METABOLIC_PROCESS | 315 | 10 | Up | 9.221e-05 | 0.004724 |
| GO_NUCLEAR_BODY | 649 | 21 | Down | 0.0001011 | 0.004828 |
| GO_MONOCARBOXYLIC_ACID_TRANSPORT | 110 | 6 | Up | 0.0001017 | 0.004829 |
| GO_CARBOHYDRATE_BIOSYNTHETIC_PROCESS | 148 | 6 | Up | 0.0001221 | 0.005132 |
| GO_SYMPORTER_ACTIVITY | 94 | 10 | Up | 0.0001224 | 0.005132 |
| GO_LIPID_BIOSYNTHETIC_PROCESS | 508 | 13 | Up | 0.0001267 | 0.005207 |
| GO_DRUG_TRANSPORT | 152 | 8 | Up | 0.0001461 | 0.005379 |
| GO_AMINO_ACID_TRANSMEMBRANE_TRANSPORT | 69 | 5 | Up | 0.0001709 | 0.005806 |
| GO_ORGANIC_ACID_TRANSMEMBRANE_TRANSPORT | 106 | 9 | Up | 0.0001766 | 0.005887 |
| GO_ORGANIC_ANION_TRANSPORT | 345 | 16 | Up | 0.0001785 | 0.005928 |
| GO_SECONDARY_ACTIVE_TRANSMEMBRANE_TRANSPORTER_ACTIVITY | 154 | 13 | Up | 0.0001819 | 0.005967 |
| GO_COENZYME_METABOLIC_PROCESS | 195 | 5 | Up | 0.000182 | 0.005967 |
| GO_ANION_TRANSMEMBRANE_TRANSPORT | 203 | 15 | Up | 0.0001854 | 0.005999 |
| GO_ANION_TRANSPORT | 434 | 22 | Up | 0.0001883 | 0.006049 |
| GO_SEQUENCE_SPECIFIC_DNA_BINDING | 827 | 29 | Down | 0.0001932 | 0.006093 |
| GO_POSITIVE_REGULATION_OF_TRANSCRIPTION_BY_RNA_POLYMERASE_II | 903 | 31 | Down | 0.000212 | 0.006139 |
| GO_REGULATORY_REGION_NUCLEIC_ACID_BINDING | 720 | 24 | Down | 0.0002154 | 0.006157 |
| GO_PROTEIN_LOCALIZATION_TO_MEMBRANE | 539 | 35 | Up | 0.0002254 | 0.006217 |
| GO_PROTEIN_METHYLATION | 153 | 8 | Down | 0.0002353 | 0.006313 |
| GO_REGULATION_OF_PHOSPHATASE_ACTIVITY | 149 | 8 | Down | 0.0002413 | 0.006313 |
| GO_ACTIVATING_TRANSCRIPTION_FACTOR_BINDING | 70 | 5 | Down | 0.0002415 | 0.006313 |
| GO_ORGANIC_ANION_TRANSMEMBRANE_TRANSPORTER_ACTIVITY | 144 | 11 | Up | 0.000253 | 0.006397 |
| GO_ESTABLISHMENT_OF_PROTEIN_LOCALIZATION_TO_MEMBRANE | 287 | 30 | Up | 0.0002603 | 0.006429 |
| GO_PROTEIN_ACETYLATION | 168 | 5 | Down | 0.0002605 | 0.006429 |
| GO_ISOPRENOID_METABOLIC_PROCESS | 68 | 5 | Up | 0.0002633 | 0.006433 |
| GO_PROTEIN_MATURATION | 210 | 8 | Up | 0.0002641 | 0.006433 |
| GO_DRUG_TRANSMEMBRANE_TRANSPORT | 65 | 6 | Up | 0.0002665 | 0.006433 |
| GO_REGULATION_OF_PEPTIDE_HORMONE_SECRETION | 157 | 9 | Up | 0.0002996 | 0.00691 |
| GO_ANION_TRANSMEMBRANE_TRANSPORTER_ACTIVITY | 228 | 17 | Up | 0.0003288 | 0.007143 |
| GO_NUCLEAR_SPECK | 346 | 11 | Down | 0.0003295 | 0.007143 |
| GO_PROXIMAL_PROMOTER_SEQUENCE_SPECIFIC_DNA_BINDING | 413 | 15 | Down | 0.0003484 | 0.007241 |
| GO_REGULATION_OF_DEPHOSPHORYLATION | 180 | 8 | Down | 0.0003661 | 0.007384 |
| GO_COVALENT_CHROMATIN_MODIFICATION | 384 | 13 | Down | 0.0003797 | 0.007483 |
| GO_REGULATION_OF_CHROMATIN_ORGANIZATION | 158 | 7 | Down | 0.0003848 | 0.007483 |
| GO_REGULATION_OF_INSULIN_SECRETION | 140 | 9 | Up | 0.0004112 | 0.007729 |
| GO_ROUGH_ENDOPLASMIC_RETICULUM | 67 | 6 | Up | 0.0004221 | 0.00781 |
| GO_AMINO_ACID_TRANSPORT | 119 | 6 | Up | 0.0004254 | 0.00781 |
| GO_SEQUENCE_SPECIFIC_DOUBLE_STRANDED_DNA_BINDING | 668 | 23 | Down | 0.0004287 | 0.007824 |
| GO_DOUBLE_STRANDED_DNA_BINDING | 740 | 30 | Down | 0.0004372 | 0.007859 |
| GO_REGULATION_OF_MRNA_PROCESSING | 117 | 5 | Down | 0.0004506 | 0.00795 |
| GO_NUCLEOSIDE_PHOSPHATE_BIOSYNTHETIC_PROCESS | 227 | 10 | Up | 0.0004663 | 0.008076 |
| GO_NUCLEOPLASM_PART | 943 | 31 | Down | 0.0004697 | 0.008088 |
| GO_SECRETORY_VESICLE | 713 | 24 | Up | 0.0004815 | 0.008218 |
| GO_AMINO_ACID_TRANSMEMBRANE_TRANSPORTER_ACTIVITY | 60 | 5 | Up | 0.000498 | 0.008306 |
| GO_PURINE_CONTAINING_COMPOUND_BIOSYNTHETIC_PROCESS | 170 | 6 | Up | 0.0004995 | 0.008306 |
| GO_LIPID_CATABOLIC_PROCESS | 221 | 7 | Up | 0.0005363 | 0.008614 |
| GO_INORGANIC_ANION_TRANSPORT | 117 | 7 | Up | 0.0005584 | 0.008701 |
| GO_MONOVALENT_INORGANIC_CATION_TRANSMEMBRANE_TRANSPORTER_ACTIVITY | 268 | 13 | Up | 0.0005832 | 0.00893 |
| GO_PEPTIDE_HORMONE_SECRETION | 194 | 9 | Up | 0.0005972 | 0.009027 |
| GO_ALPHA_AMINO_ACID_METABOLIC_PROCESS | 126 | 6 | Up | 0.0006074 | 0.009142 |
| GO_ORGANIC_ACID_CATABOLIC_PROCESS | 184 | 5 | Up | 0.0006398 | 0.009283 |
| GO_IMPORT_INTO_CELL | 532 | 7 | Up | 0.0006709 | 0.009508 |
| GO_RIBOSE_PHOSPHATE_BIOSYNTHETIC_PROCESS | 165 | 7 | Up | 0.0006731 | 0.009508 |
| GO_TUBULIN_BINDING | 258 | 5 | Down | 0.0006736 | 0.009508 |
| GO_CATION_TRANSMEMBRANE_TRANSPORTER_ACTIVITY | 458 | 18 | Up | 0.0006766 | 0.009508 |
| GO_MRNA_METABOLIC_PROCESS | 453 | 26 | Down | 0.0006959 | 0.009529 |
| GO_GUANYL_NUCLEOTIDE_BINDING | 317 | 6 | Up | 0.0007188 | 0.009678 |
| GO_ORGANIC_HYDROXY_COMPOUND_BIOSYNTHETIC_PROCESS | 183 | 8 | Up | 0.000736 | 0.00984 |
Exploration of Significant GO Gene Sets
goFry %>%
left_join(gsSizes) %>%
left_join(distinct(go, gs_name, shortest_path)) %>%
mutate(
propDE = de_size / gs_size
) %>%
dplyr::filter(
de_size >= 15,
propDE > 0.05,
FDR < 0.02,
shortest_path > 3
) %>%
dplyr::select(gs_name, de_id) %>%
unnest(de_id) %>%
mutate(
gs_name = str_remove(gs_name, "GO_"),
gs_name = fct_lump(gs_name, n = 20)
) %>%
split(f = .$gs_name) %>%
lapply(magrittr::extract2, "de_id") %>%
fromList() %>%
upset(
nsets = length(.),
nintersects = 20,
order.by = "freq",
mb.ratio = c(0.6, 0.4),
sets.x.label = "Number Of DE Genes"
)
UpSet plot indicating distribution of DE genes within significantly enriched terms from the GO gene sets. For this visualisation, GO terms were restricted to those with 15 or more DE genes, where this represented more than 5% of a gene set as being DE, along with an FDR < 0.02 and more than 3 steps back to the ontology root. The 20 largest GO terms satisfying these criteria are shown. The plot is truncated at the right hand side for simplicity. A group of 28 genes is uniquely attributed to the Mitochondrial Envelope, with a further 18 being relatively unique to mRNA Metabolic Process. The next grouping of 15 genes are unique to Regulation Of Nucleobase-Containing Compound Metabolic Process followed by 25 genes, spread across two clusters of terms which largely represent Ribosomal activity. In between these are 13 genes uniquely associated with Anion Transport.
| Version | Author | Date |
|---|---|---|
| a5cdf5d | Steve Ped | 2020-03-06 |
Genes Associated with the Mitochondrial Envelope
go %>%
dplyr::filter(
gs_name == "GO_MITOCHONDRIAL_ENVELOPE",
gene_id %in% dplyr::filter(deTable, DE)$gene_id
) %>%
dplyr::select(
gene_id, gene_name
) %>%
left_join(
cpmPostNorm %>%
as.data.frame() %>%
rownames_to_column("gene_id")
) %>%
dplyr::select(gene_name, starts_with("Ps")) %>%
as.data.frame() %>%
column_to_rownames("gene_name") %>%
as.matrix() %>%
pheatmap(
color = viridis_pal(option = "magma")(100),
legend_breaks = c(seq(-2, 8, by = 2), max(.)),
legend_labels = c(seq(-2, 8, by = 2), "logCPM\n"),
labels_col = hmHeat %>%
dplyr::select(starts_with("Ps")) %>%
colnames() %>%
enframe(name = NULL) %>%
dplyr::rename(sample = value) %>%
left_join(dgeList$samples) %>%
dplyr::select(sample, sampleID) %>%
with(
structure(sampleID, names = sample)
),
annotation_col = dgeList$samples %>%
dplyr::select(Genotype = genotype),
cutree_rows = 4,
cutree_cols = 2
)
All DE genes associated with the GO term ‘Mitochondrial Envelope’.
| Version | Author | Date |
|---|---|---|
| a5cdf5d | Steve Ped | 2020-03-06 |
Genes Associated with RNA Metabolic Process
go %>%
dplyr::filter(
gs_name == "GO_MRNA_METABOLIC_PROCESS",
gene_id %in% dplyr::filter(deTable, DE)$gene_id
) %>%
dplyr::select(
gene_id, gene_name
) %>%
left_join(
cpmPostNorm %>%
as.data.frame() %>%
rownames_to_column("gene_id")
) %>%
dplyr::select(gene_name, starts_with("Ps")) %>%
as.data.frame() %>%
column_to_rownames("gene_name") %>%
as.matrix() %>%
pheatmap(
color = viridis_pal(option = "magma")(100),
legend_breaks = c(seq(-2, 8, by = 2), max(.)),
legend_labels = c(seq(-2, 8, by = 2), "logCPM\n"),
labels_col = hmHeat %>%
dplyr::select(starts_with("Ps")) %>%
colnames() %>%
enframe(name = NULL) %>%
dplyr::rename(sample = value) %>%
left_join(dgeList$samples) %>%
dplyr::select(sample, sampleID) %>%
with(
structure(sampleID, names = sample)
),
annotation_col = dgeList$samples %>%
dplyr::select(Genotype = genotype),
cutree_rows = 4,
cutree_cols = 2
)
All DE genes associated with the GO term ‘mRNA Metabolic Process’.
| Version | Author | Date |
|---|---|---|
| a5cdf5d | Steve Ped | 2020-03-06 |
Genes Associated with Regulation Of Nucleobase-Containing Compound Metabolic Process
go %>%
dplyr::filter(
gs_name == "GO_REGULATION_OF_NUCLEOBASE_CONTAINING_COMPOUND_METABOLIC_PROCESS",
gene_id %in% dplyr::filter(deTable, DE)$gene_id
) %>%
dplyr::select(
gene_id, gene_name
) %>%
left_join(
cpmPostNorm %>%
as.data.frame() %>%
rownames_to_column("gene_id")
) %>%
dplyr::select(gene_name, starts_with("Ps")) %>%
as.data.frame() %>%
column_to_rownames("gene_name") %>%
as.matrix() %>%
pheatmap(
color = viridis_pal(option = "magma")(100),
legend_breaks = c(seq(-2, 8, by = 2), max(.)),
legend_labels = c(seq(-2, 8, by = 2), "logCPM\n"),
labels_col = hmHeat %>%
dplyr::select(starts_with("Ps")) %>%
colnames() %>%
enframe(name = NULL) %>%
dplyr::rename(sample = value) %>%
left_join(dgeList$samples) %>%
dplyr::select(sample, sampleID) %>%
with(
structure(sampleID, names = sample)
),
annotation_col = dgeList$samples %>%
dplyr::select(Genotype = genotype),
cutree_rows = 4,
cutree_cols = 2
)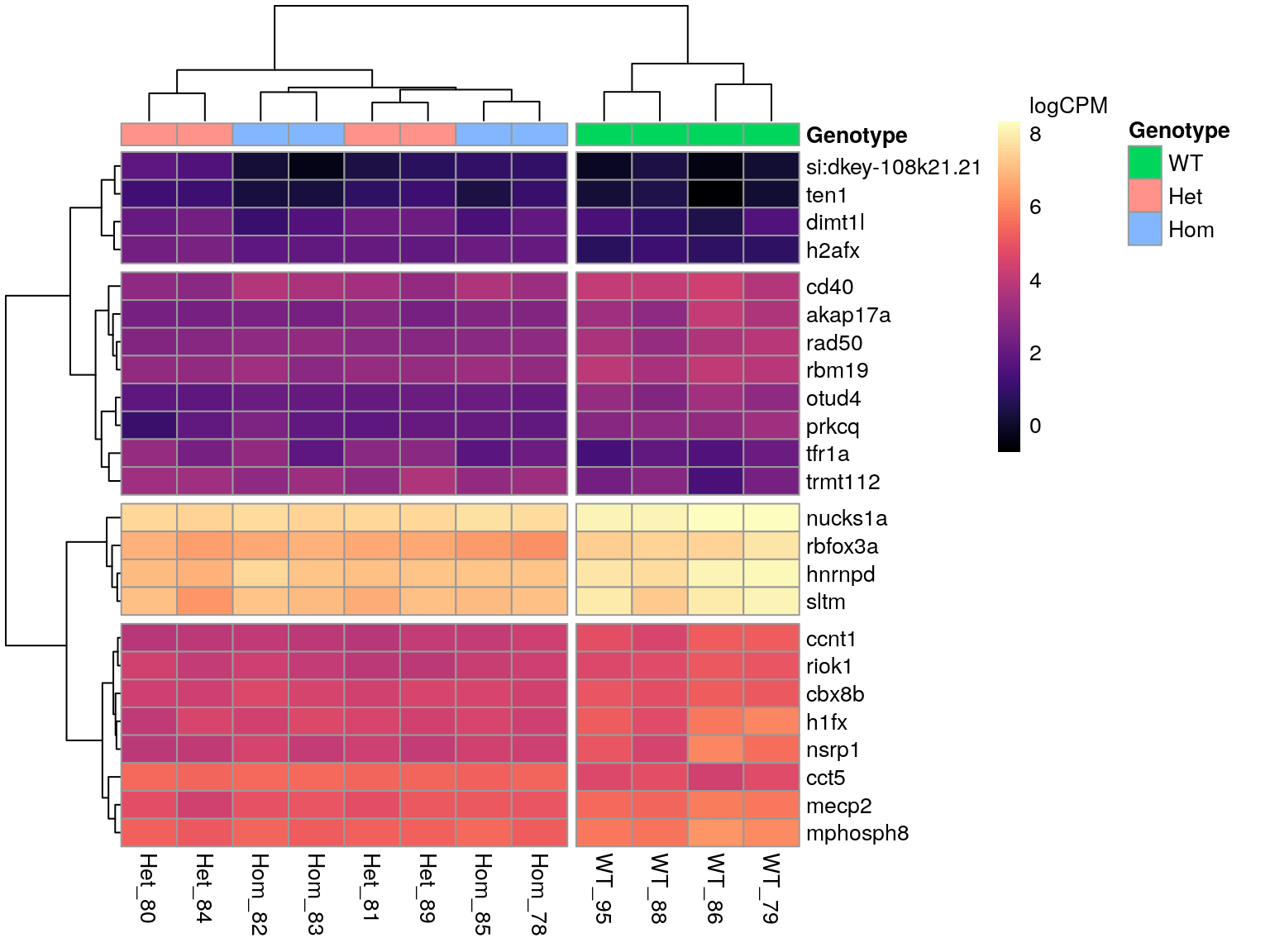
All DE genes associated with the GO term ‘mRNA Metabolic Process’.
| Version | Author | Date |
|---|---|---|
| a5cdf5d | Steve Ped | 2020-03-06 |
Genes Associated with Anion Transport
go %>%
dplyr::filter(
gs_name == "GO_ANION_TRANSPORT",
gene_id %in% dplyr::filter(deTable, DE)$gene_id
) %>%
dplyr::select(
gene_id, gene_name
) %>%
left_join(
cpmPostNorm %>%
as.data.frame() %>%
rownames_to_column("gene_id")
) %>%
dplyr::select(gene_name, starts_with("Ps")) %>%
as.data.frame() %>%
column_to_rownames("gene_name") %>%
as.matrix() %>%
pheatmap(
color = viridis_pal(option = "magma")(100),
legend_breaks = c(seq(-2, 8, by = 2), max(.)),
legend_labels = c(seq(-2, 8, by = 2), "logCPM\n"),
labels_col = hmHeat %>%
dplyr::select(starts_with("Ps")) %>%
colnames() %>%
enframe(name = NULL) %>%
dplyr::rename(sample = value) %>%
left_join(dgeList$samples) %>%
dplyr::select(sample, sampleID) %>%
with(
structure(sampleID, names = sample)
),
annotation_col = dgeList$samples %>%
dplyr::select(Genotype = genotype),
cutree_rows = 3,
cutree_cols = 2
)
All DE genes associated with the GO term ‘Anion Transport’.
| Version | Author | Date |
|---|---|---|
| a5cdf5d | Steve Ped | 2020-03-06 |
Genes Associated with Ribosomal Activity
go %>%
dplyr::filter(
gs_name == "GO_CYTOSOLIC_RIBOSOME",
gene_id %in% dplyr::filter(deTable, DE)$gene_id
) %>%
dplyr::select(
gene_id, gene_name
) %>%
left_join(
cpmPostNorm %>%
as.data.frame() %>%
rownames_to_column("gene_id")
) %>%
dplyr::select(gene_name, starts_with("Ps")) %>%
as.data.frame() %>%
column_to_rownames("gene_name") %>%
as.matrix() %>%
pheatmap(
color = viridis_pal(option = "magma")(100),
legend_breaks = c(seq(-2, 8, by = 2), max(.)),
legend_labels = c(seq(-2, 8, by = 2), "logCPM\n"),
labels_col = hmHeat %>%
dplyr::select(starts_with("Ps")) %>%
colnames() %>%
enframe(name = NULL) %>%
dplyr::rename(sample = value) %>%
left_join(dgeList$samples) %>%
dplyr::select(sample, sampleID) %>%
with(
structure(sampleID, names = sample)
),
annotation_col = dgeList$samples %>%
dplyr::select(Genotype = genotype),
cutree_rows = 3,
cutree_cols = 2
)
All DE genes associated with the GO term ‘Cytosolic Ribosome’.
| Version | Author | Date |
|---|---|---|
| a5cdf5d | Steve Ped | 2020-03-06 |
Data Export
All enriched gene sets with an FDR adjusted p-value < 0.05 were exported as a single csv file.
bind_rows(
hmFry,
kgFry,
goFry
) %>%
dplyr::filter(FDR < 0.05) %>%
left_join(gsSizes) %>%
dplyr::select(
gs_name, NGenes, Direction, PValue, FDR,
gene_symbol, de_size
) %>%
mutate(
DE = lapply(
X = gene_symbol,
FUN = intersect,
y = dplyr::filter(deTable, DE)$gene_name
),
DE = vapply(DE, paste, character(1), collapse = ";")
) %>%
dplyr::select(
`Gene Set` = gs_name,
`Nbr Detected Genes` = NGenes,
`Nbr DE Genes` = de_size,
Direction, PValue, FDR,
`DE Genes` = DE
) %>%
write_csv(
here::here("output", "Enrichment_Mutant_V_WT.csv")
)
devtools::session_info()─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 3.6.3 (2020-02-29)
os Ubuntu 18.04.4 LTS
system x86_64, linux-gnu
ui X11
language en_AU:en
collate en_AU.UTF-8
ctype en_AU.UTF-8
tz Australia/Melbourne
date 2020-03-07
─ Packages ───────────────────────────────────────────────────────────────────
package * version date lib source
AnnotationDbi * 1.48.0 2019-10-29 [2] Bioconductor
askpass 1.1 2019-01-13 [2] CRAN (R 3.6.0)
assertthat 0.2.1 2019-03-21 [2] CRAN (R 3.6.0)
backports 1.1.5 2019-10-02 [2] CRAN (R 3.6.1)
BiasedUrn * 1.07 2015-12-28 [2] CRAN (R 3.6.1)
Biobase * 2.46.0 2019-10-29 [2] Bioconductor
BiocFileCache 1.10.2 2019-11-08 [2] Bioconductor
BiocGenerics * 0.32.0 2019-10-29 [2] Bioconductor
BiocParallel 1.20.1 2019-12-21 [2] Bioconductor
biomaRt 2.42.0 2019-10-29 [2] Bioconductor
Biostrings 2.54.0 2019-10-29 [2] Bioconductor
bit 1.1-15.2 2020-02-10 [2] CRAN (R 3.6.2)
bit64 0.9-7 2017-05-08 [2] CRAN (R 3.6.0)
bitops 1.0-6 2013-08-17 [2] CRAN (R 3.6.0)
blob 1.2.1 2020-01-20 [2] CRAN (R 3.6.2)
broom 0.5.4 2020-01-27 [2] CRAN (R 3.6.2)
callr 3.4.2 2020-02-12 [2] CRAN (R 3.6.2)
cellranger 1.1.0 2016-07-27 [2] CRAN (R 3.6.0)
cli 2.0.1 2020-01-08 [2] CRAN (R 3.6.2)
cluster 2.1.0 2019-06-19 [2] CRAN (R 3.6.1)
colorspace 1.4-1 2019-03-18 [2] CRAN (R 3.6.0)
crayon 1.3.4 2017-09-16 [2] CRAN (R 3.6.0)
curl 4.3 2019-12-02 [2] CRAN (R 3.6.2)
data.table 1.12.8 2019-12-09 [2] CRAN (R 3.6.2)
DBI 1.1.0 2019-12-15 [2] CRAN (R 3.6.2)
dbplyr 1.4.2 2019-06-17 [2] CRAN (R 3.6.0)
DelayedArray 0.12.2 2020-01-06 [2] Bioconductor
desc 1.2.0 2018-05-01 [2] CRAN (R 3.6.0)
devtools 2.2.2 2020-02-17 [2] CRAN (R 3.6.2)
digest 0.6.25 2020-02-23 [2] CRAN (R 3.6.2)
dplyr * 0.8.4 2020-01-31 [2] CRAN (R 3.6.2)
edgeR * 3.28.1 2020-02-26 [2] Bioconductor
ellipsis 0.3.0 2019-09-20 [2] CRAN (R 3.6.1)
evaluate 0.14 2019-05-28 [2] CRAN (R 3.6.0)
FactoMineR 2.2 2020-02-05 [2] CRAN (R 3.6.2)
fansi 0.4.1 2020-01-08 [2] CRAN (R 3.6.2)
farver 2.0.3 2020-01-16 [2] CRAN (R 3.6.2)
flashClust 1.01-2 2012-08-21 [2] CRAN (R 3.6.1)
forcats * 0.4.0 2019-02-17 [2] CRAN (R 3.6.0)
fs 1.3.1 2019-05-06 [2] CRAN (R 3.6.0)
geneLenDataBase * 1.22.0 2019-11-05 [2] Bioconductor
generics 0.0.2 2018-11-29 [2] CRAN (R 3.6.0)
GenomeInfoDb 1.22.0 2019-10-29 [2] Bioconductor
GenomeInfoDbData 1.2.2 2019-11-21 [2] Bioconductor
GenomicAlignments 1.22.1 2019-11-12 [2] Bioconductor
GenomicFeatures 1.38.2 2020-02-15 [2] Bioconductor
GenomicRanges 1.38.0 2019-10-29 [2] Bioconductor
ggdendro 0.1-20 2016-04-27 [2] CRAN (R 3.6.0)
ggplot2 * 3.2.1 2019-08-10 [2] CRAN (R 3.6.1)
ggrepel 0.8.1 2019-05-07 [2] CRAN (R 3.6.0)
git2r 0.26.1 2019-06-29 [2] CRAN (R 3.6.1)
glue 1.3.1 2019-03-12 [2] CRAN (R 3.6.0)
GO.db 3.10.0 2019-11-21 [2] Bioconductor
goseq * 1.38.0 2019-10-29 [2] Bioconductor
gridExtra 2.3 2017-09-09 [2] CRAN (R 3.6.0)
gtable 0.3.0 2019-03-25 [2] CRAN (R 3.6.0)
haven 2.2.0 2019-11-08 [2] CRAN (R 3.6.1)
here 0.1 2017-05-28 [2] CRAN (R 3.6.0)
highr 0.8 2019-03-20 [2] CRAN (R 3.6.0)
hms 0.5.3 2020-01-08 [2] CRAN (R 3.6.2)
htmltools 0.4.0 2019-10-04 [2] CRAN (R 3.6.1)
htmlwidgets 1.5.1 2019-10-08 [2] CRAN (R 3.6.1)
httpuv 1.5.2 2019-09-11 [2] CRAN (R 3.6.1)
httr 1.4.1 2019-08-05 [2] CRAN (R 3.6.1)
hwriter 1.3.2 2014-09-10 [2] CRAN (R 3.6.0)
IRanges * 2.20.2 2020-01-13 [2] Bioconductor
jpeg 0.1-8.1 2019-10-24 [2] CRAN (R 3.6.1)
jsonlite 1.6.1 2020-02-02 [2] CRAN (R 3.6.2)
kableExtra 1.1.0 2019-03-16 [2] CRAN (R 3.6.1)
knitr 1.28 2020-02-06 [2] CRAN (R 3.6.2)
labeling 0.3 2014-08-23 [2] CRAN (R 3.6.0)
later 1.0.0 2019-10-04 [2] CRAN (R 3.6.1)
lattice 0.20-40 2020-02-19 [4] CRAN (R 3.6.2)
latticeExtra 0.6-29 2019-12-19 [2] CRAN (R 3.6.2)
lazyeval 0.2.2 2019-03-15 [2] CRAN (R 3.6.0)
leaps 3.1 2020-01-16 [2] CRAN (R 3.6.2)
lifecycle 0.1.0 2019-08-01 [2] CRAN (R 3.6.1)
limma * 3.42.2 2020-02-03 [2] Bioconductor
locfit 1.5-9.1 2013-04-20 [2] CRAN (R 3.6.0)
lubridate 1.7.4 2018-04-11 [2] CRAN (R 3.6.0)
magrittr * 1.5 2014-11-22 [2] CRAN (R 3.6.0)
MASS 7.3-51.5 2019-12-20 [4] CRAN (R 3.6.2)
Matrix 1.2-18 2019-11-27 [2] CRAN (R 3.6.1)
matrixStats 0.55.0 2019-09-07 [2] CRAN (R 3.6.1)
memoise 1.1.0 2017-04-21 [2] CRAN (R 3.6.0)
mgcv 1.8-31 2019-11-09 [4] CRAN (R 3.6.1)
modelr 0.1.6 2020-02-22 [2] CRAN (R 3.6.2)
msigdbr * 7.0.1 2019-09-04 [2] CRAN (R 3.6.2)
munsell 0.5.0 2018-06-12 [2] CRAN (R 3.6.0)
ngsReports * 1.1.2 2019-10-16 [1] Bioconductor
nlme 3.1-144 2020-02-06 [4] CRAN (R 3.6.2)
openssl 1.4.1 2019-07-18 [2] CRAN (R 3.6.1)
pander * 0.6.3 2018-11-06 [2] CRAN (R 3.6.0)
pheatmap * 1.0.12 2019-01-04 [2] CRAN (R 3.6.0)
pillar 1.4.3 2019-12-20 [2] CRAN (R 3.6.2)
pkgbuild 1.0.6 2019-10-09 [2] CRAN (R 3.6.1)
pkgconfig 2.0.3 2019-09-22 [2] CRAN (R 3.6.1)
pkgload 1.0.2 2018-10-29 [2] CRAN (R 3.6.0)
plotly 4.9.2 2020-02-12 [2] CRAN (R 3.6.2)
plyr 1.8.5 2019-12-10 [2] CRAN (R 3.6.2)
png 0.1-7 2013-12-03 [2] CRAN (R 3.6.0)
prettyunits 1.1.1 2020-01-24 [2] CRAN (R 3.6.2)
processx 3.4.2 2020-02-09 [2] CRAN (R 3.6.2)
progress 1.2.2 2019-05-16 [2] CRAN (R 3.6.0)
promises 1.1.0 2019-10-04 [2] CRAN (R 3.6.1)
ps 1.3.2 2020-02-13 [2] CRAN (R 3.6.2)
purrr * 0.3.3 2019-10-18 [2] CRAN (R 3.6.1)
R6 2.4.1 2019-11-12 [2] CRAN (R 3.6.1)
rappdirs 0.3.1 2016-03-28 [2] CRAN (R 3.6.0)
RColorBrewer * 1.1-2 2014-12-07 [2] CRAN (R 3.6.0)
Rcpp 1.0.3 2019-11-08 [2] CRAN (R 3.6.1)
RCurl 1.98-1.1 2020-01-19 [2] CRAN (R 3.6.2)
readr * 1.3.1 2018-12-21 [2] CRAN (R 3.6.0)
readxl 1.3.1 2019-03-13 [2] CRAN (R 3.6.0)
remotes 2.1.1 2020-02-15 [2] CRAN (R 3.6.2)
reprex 0.3.0 2019-05-16 [2] CRAN (R 3.6.0)
reshape2 1.4.3 2017-12-11 [2] CRAN (R 3.6.0)
rlang 0.4.4 2020-01-28 [2] CRAN (R 3.6.2)
rmarkdown 2.1 2020-01-20 [2] CRAN (R 3.6.2)
rprojroot 1.3-2 2018-01-03 [2] CRAN (R 3.6.0)
Rsamtools 2.2.3 2020-02-23 [2] Bioconductor
RSQLite 2.2.0 2020-01-07 [2] CRAN (R 3.6.2)
rstudioapi 0.11 2020-02-07 [2] CRAN (R 3.6.2)
rtracklayer 1.46.0 2019-10-29 [2] Bioconductor
rvest 0.3.5 2019-11-08 [2] CRAN (R 3.6.1)
S4Vectors * 0.24.3 2020-01-18 [2] Bioconductor
scales * 1.1.0 2019-11-18 [2] CRAN (R 3.6.1)
scatterplot3d 0.3-41 2018-03-14 [2] CRAN (R 3.6.1)
sessioninfo 1.1.1 2018-11-05 [2] CRAN (R 3.6.0)
ShortRead 1.44.3 2020-02-03 [2] Bioconductor
statmod 1.4.34 2020-02-17 [2] CRAN (R 3.6.2)
stringi 1.4.6 2020-02-17 [2] CRAN (R 3.6.2)
stringr * 1.4.0 2019-02-10 [2] CRAN (R 3.6.0)
SummarizedExperiment 1.16.1 2019-12-19 [2] Bioconductor
testthat 2.3.1 2019-12-01 [2] CRAN (R 3.6.1)
tibble * 2.1.3 2019-06-06 [2] CRAN (R 3.6.0)
tidyr * 1.0.2 2020-01-24 [2] CRAN (R 3.6.2)
tidyselect 1.0.0 2020-01-27 [2] CRAN (R 3.6.2)
tidyverse * 1.3.0 2019-11-21 [2] CRAN (R 3.6.1)
truncnorm 1.0-8 2018-02-27 [2] CRAN (R 3.6.0)
UpSetR * 1.4.0 2019-05-22 [2] CRAN (R 3.6.0)
usethis 1.5.1 2019-07-04 [2] CRAN (R 3.6.1)
vctrs 0.2.3 2020-02-20 [2] CRAN (R 3.6.2)
viridisLite 0.3.0 2018-02-01 [2] CRAN (R 3.6.0)
webshot 0.5.2 2019-11-22 [2] CRAN (R 3.6.1)
whisker 0.4 2019-08-28 [2] CRAN (R 3.6.1)
withr 2.1.2 2018-03-15 [2] CRAN (R 3.6.0)
workflowr * 1.6.0 2019-12-19 [2] CRAN (R 3.6.2)
xfun 0.12 2020-01-13 [2] CRAN (R 3.6.2)
XML 3.99-0.3 2020-01-20 [2] CRAN (R 3.6.2)
xml2 1.2.2 2019-08-09 [2] CRAN (R 3.6.1)
XVector 0.26.0 2019-10-29 [2] Bioconductor
yaml 2.2.1 2020-02-01 [2] CRAN (R 3.6.2)
zlibbioc 1.32.0 2019-10-29 [2] Bioconductor
zoo 1.8-7 2020-01-10 [2] CRAN (R 3.6.2)
[1] /home/steveped/R/x86_64-pc-linux-gnu-library/3.6
[2] /usr/local/lib/R/site-library
[3] /usr/lib/R/site-library
[4] /usr/lib/R/library