Last updated: 2025-11-04
Checks: 6 1
Knit directory:
Integrating-nir-genomic-kernel/
This reproducible R Markdown analysis was created with workflowr (version 1.7.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20250829) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version df8457c. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/Article_documents/
Ignored: data/Maize-NIRS-GBS-main/
Ignored: output/Pred.ability/grafico_CV1_CV2.tiff
Ignored: output/Pred.ability/grafico_exemplo_CV0_CV00.tiff
Ignored: output/Pred.ability/grafico_exemplo_GY_CV0.tiff
Ignored: output/Pred.ability/graficos_gerados/
Ignored: output/ajuste_modelo/
Ignored: output/climate_results/combined_climate_plot.tiff
Ignored: output/componentes_variancia/Variance.Components.DAT.Means.Parsed.csv
Ignored: output/componentes_variancia/Variance.Components.DAT.Means.csv
Ignored: output/componentes_variancia/rep_1/
Ignored: output/componentes_variancia/rep_10/
Ignored: output/componentes_variancia/rep_11/
Ignored: output/componentes_variancia/rep_12/
Ignored: output/componentes_variancia/rep_13/
Ignored: output/componentes_variancia/rep_14/
Ignored: output/componentes_variancia/rep_15/
Ignored: output/componentes_variancia/rep_16/
Ignored: output/componentes_variancia/rep_17/
Ignored: output/componentes_variancia/rep_18/
Ignored: output/componentes_variancia/rep_19/
Ignored: output/componentes_variancia/rep_2/
Ignored: output/componentes_variancia/rep_20/
Ignored: output/componentes_variancia/rep_3/
Ignored: output/componentes_variancia/rep_4/
Ignored: output/componentes_variancia/rep_5/
Ignored: output/componentes_variancia/rep_6/
Ignored: output/componentes_variancia/rep_7/
Ignored: output/componentes_variancia/rep_8/
Ignored: output/componentes_variancia/rep_9/
Ignored: output/componentes_variancia/variance_components_percentage_COMBINED.tiff
Ignored: output/cv-schemes.tiff
Ignored: output/results/
Unstaged changes:
Modified: analysis/analysis.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/analysis.Rmd) and HTML
(docs/analysis.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | df8457c | WevertonGomesCosta | 2025-11-04 | update analisys.Rmd and .html |
| html | df8457c | WevertonGomesCosta | 2025-11-04 | update analisys.Rmd and .html |
| Rmd | 1cbf0a0 | WevertonGomesCosta | 2025-11-03 | update analysis .rmd and .html |
| html | 1cbf0a0 | WevertonGomesCosta | 2025-11-03 | update analysis .rmd and .html |
| Rmd | 2dac00d | WevertonGomesCosta | 2025-11-03 | add analysis .rmd and html |
| html | 2dac00d | WevertonGomesCosta | 2025-11-03 | add analysis .rmd and html |
| Rmd | 904b83c | WevertonGomesCosta | 2025-11-03 | Remove arquivos de output antigos |
| Rmd | fe31a33 | WevertonGomesCosta | 2025-11-03 | add analysis.rmd script |
Analysis of NIR and Genomic Kernel Integration
Este documento detalha o pipeline de análise para integrar dados de Espectroscopia de Refletância no Infravermelho Próximo (NIR), dados genômicos e climáticos usando métodos de kernel para aumentar a precisão da predição. A análise inclui o pré-processamento de dados, ajuste de modelos e avaliação do desempenho preditivo.
Este script foca especificamente nas etapas iniciais e cruciais de carregamento, limpeza e alinhamento dos diferentes tipos de dados.
1) Load Required Libraries and Data
Nesta etapa, carregamos nosso “kit de ferramentas” de pacotes R. Cada um deles é essencial para diferentes partes da análise:
tidyverse: Uma coleção de pacotes (comodplyretidyr) para a manipulação, limpeza e organização geral dos dados.data.table: Extremamente eficiente para ler e manipular arquivos de dados muito grandes, como nossas matrizes genômicas (SNPs) e espectrais (NIR).BGLR: O pacote central para a modelagem Bayesiana. Ele será usado posteriormente para ajustar os modelos de predição genômica usando os kernels que prepararemos.parallel&doParallel: Ferramentas para paralelizar nossos cálculos. Essenciais para rodar as análises de validação cruzada (CV) em tempo hábil.MASS: Contém funções estatísticas adicionais que podem ser úteis.
# Pacotes principais
library(tidyverse) # Manipulação de dados
library(data.table) # Leitura eficiente de grandes arquivos
library(BGLR) # Modelagem Bayesiana para predição genômica
# Paralelismo
library(parallel)
library(doParallel)
# Funções estatísticas adicionais
library(MASS) # Funções estatísticas
library(gridExtra)
library(grid) 2) Data Loading and Preprocessing
Os dados utilizados nesta análise vêm de três fontes principais. O objetivo desta seção é carregá-los e garantir que estejam consistentes entre si.
2.1) Carregamento dos Dados
Primeiro, carregamos os arquivos brutos do disco.
- NIR.csv: Este é um arquivo rico que contém
múltiplos tipos de informação:
- IDs (
Pedigree,Env). - As variáveis de resposta (traits) que queremos prever, como
GY(Produtividade) eKW(Peso de Grão), que provavelmente são BLUPs calculados anteriormente. - As próprias leituras espectrais (bandas do NIR), que usaremos como preditores.
- IDs (
- GAPIT.Genotype.Numerical.txt: Contém os dados
genotípicos.
taxa: O ID do genótipo.- Colunas de marcadores (SNPs) em formato numérico (ex: -1, 0, 1), que é o formato ideal para construir uma matriz de parentesco genômico (Kernel G).
- Pheno: Este objeto não é carregado de um arquivo.
Em vez disso, ele é extraído do arquivo
NIRpara isolar as variáveis de resposta (fenótipos) que nossos modelos tentarão prever.
Dados fenotípicos com leituras espectrais e os blups
# Carrega dados fenômicos (NIR)
NIR <- fread("data/NIR.csv") %>% as.data.frame()
# Visualização inicial
head(NIR[, 1:7]) # primeiras colunasunique(NIR$Env) # ambientes disponíveis[1] "CS11_WS" "CS11_WW" "CS12_WS" "CS12_WW"Dados genotípicos com marcadores SNP
# Carrega dados genotípicos (SNPs processados pelo GAPIT)
Geno <- fread("data/GAPIT.Genotype.Numerical.txt") %>% as.data.frame()
# Visualização inicial
head(Geno[, 1:5])Dados fenotipicos com BLUPs
# Extrai as colunas de ID (Pedigree, Env) e as traits (GY, KW) do
# dataframe NIR para criar nosso dataframe 'Pheno' (fenótipos).
Pheno <- NIR[, c("Pedigree", "Env", "GY", "KW")] %>%
arrange(Env, Pedigree) # Ordena os dados para consistência
# Visualização inicial
head(Pheno)2.2) Alinhamento de Genótipos
Esta é uma etapa crítica. Nossos modelos assumem que
a linha i da matriz genômica (Geno) corresponde ao
indivíduo da linha i da matriz fenotípica (Pheno).
Se os dados não estiverem perfeitamente alinhados, os resultados serão
inválidos.
Aqui, garantimos que todos os dataframes (NIR,
Geno, Pheno) contenham exatamente o
mesmo conjunto de genótipos.
Filtra os dados para manter apenas os genótipos compartilhados em ambos os conjuntos
# Identifica genótipos compartilhados
# A função 'intersect' encontra os IDs de genótipos (Pedigree/taxa)
# que estão presentes EM AMBOS os arquivos, Geno e NIR.
Pedigree <- intersect(Geno$taxa, NIR$Pedigree)
# Filtra os dataframes para manter APENAS os genótipos compartilhados
NIR <- NIR[NIR$Pedigree %in% Pedigree, ]
Geno <- Geno[Geno$taxa %in% Pedigree, ]
Pheno <- Pheno[Pheno$Pedigree %in% Pedigree, ]
# Checagem de sanidade: verifica se o número de genótipos únicos
# é o mesmo em todos os objetos de dados.
cat("Genótipos em NIR:", length(unique(NIR$Pedigree)), "\n")Genótipos em NIR: 329 cat("Genótipos em Geno:", length(Geno$taxa), "\n")Genótipos em Geno: 329 cat("Genótipos em Pheno:", length(unique(Pheno$Pedigree)), "\n")Genótipos em Pheno: 329 3) Dividindo os dados NIR por ambiente
Para modelar as interações Genótipo-por-Ambiente (GxE), muitas vezes precisamos analisar os dados por ambiente ou construir kernels específicos para cada ambiente.
Esta seção prepara os dados para essa análise, separando o
dataframe NIR (que contém os dados espectrais) em
quatro subconjuntos, um para cada ambiente experimental.
Separa NIR em quatro subconjuntos (a, b, c, d), correspondentes aos ambientes CS11_WS, CS11_WW, CS12_WS e CS12_WW.
# Cria um dataframe separado para cada um dos quatro ambientes
a <- NIR[NIR$Env == "CS11_WS", ]
b <- NIR[NIR$Env == "CS11_WW", ]
c <- NIR[NIR$Env == "CS12_WS", ]
d <- NIR[NIR$Env == "CS12_WW", ]
# Define nomes de linhas
# Define o 'Pedigree' como o nome da linha (rownames).
# Isso é essencial para alinhar matrizes e kernels posteriormente.
rownames(a) <- a$Pedigree
rownames(b) <- b$Pedigree
rownames(c) <- c$Pedigree
rownames(d) <- d$Pedigree
# Ordena por Pedigree
# Garante que as linhas em cada dataframe ambiental estejam
# na mesma ordem alfabética de 'Pedigree'.
a <- a[order(rownames(a)), ]
b <- b[order(rownames(b)), ]
c <- c[order(rownames(c)), ]
d <- d[order(rownames(d)), ]3.1) Identificação de Genótipos Comuns a Todos os Ambientes
Embora já tenhamos filtrado os genótipos comuns entre
Geno e NIR, alguns genótipos podem não ter
sido cultivados em todos os quatro ambientes.
Para análises de interação GxE que exigem dados balanceados (ou para criar a matriz de parentesco principal), geralmente usamos apenas o conjunto de genótipos que têm dados em todos os ambientes.
Identifica os genótipos presentes em todos os ambientes
# Identifica os genótipos presentes em todos os ambientes
# 'Reduce(intersect, ...)' aplica a função 'intersect' cumulativamente
# à lista de IDs de pedigree dos 4 ambientes (a, b, c, d) e do 'Geno'.
# O resultado é a lista de genótipos que estão presentes em TODO LUGAR.
Pedigree_communs <- Reduce(intersect, list(Geno$taxa, a$Pedigree, b$Pedigree, c$Pedigree, d$Pedigree))
length(Pedigree_communs) # Número de genótipos consistentes[1] 1464) Criando a matriz de relacionamento genômico (ZG)
Nesta seção, criamos o kernel genômico, ZG. Um
“kernel” é fundamentalmente uma matriz de parentesco ou
similaridade.
O passo crucial aqui é que, em vez de criar um kernel único para os 300+ genótipos únicos, estamos criando um kernel para as observações (genótipo dentro de um ambiente). A lógica é:
- Ordenar e Preparar
Geno: Asseguramos que a matriz genômica (Geno) esteja ordenada alfabeticamente por genótipo (taxa), definimos osrownamese removemos a coluna de ID, deixando apenas os marcadores numéricos. - Escalonar Por Ambiente: Este é o passo
mais importante. Usamos
scale(subset(Geno, ...))para padronizar (centralizar e escalonar) os marcadores SNP. Note que isso é feito separadamente para o conjunto de genótipos em cada ambiente (a, b, c, d). Isso padroniza os alelos com base na média e variância daquele subconjunto específico de indivíduos. - Empilhar (
rbind): Combinamos as quatro matrizes genômicas escalonadas (Geno.a, Geno.b, Geno.c, Geno.d) em uma única matriz grande,Geno.all. A ordem das linhas emGeno.allagora corresponde à ordem das observações no dataframePheno. - Calcular o Kernel (
tcrossprod): Usamos o produto cruzado (tcrossprod) dividido pelo número de marcadores (ncol) para calcular a matriz de parentesco genômico (similar ao método 1 de VanRaden).
O resultado, ZG, é uma grande matriz de parentesco (N
total de observações x N total de observações) que representa a
similaridade genômica entre todas as observações.
# Ordena os dados genotípicos por taxa
Geno <- Geno[order(Geno$taxa), ]
rownames(Geno) <- Geno$taxa
Geno <- Geno[, -1] # remove coluna taxa
# Escalonamento por ambiente
Geno.a <- scale(subset(Geno, rownames(Geno) %in% a$Pedigree), center = TRUE, scale = TRUE)
Geno.b <- scale(subset(Geno, rownames(Geno) %in% b$Pedigree), center = TRUE, scale = TRUE)
Geno.c <- scale(subset(Geno, rownames(Geno) %in% c$Pedigree), center = TRUE, scale = TRUE)
Geno.d <- scale(subset(Geno, rownames(Geno) %in% d$Pedigree), center = TRUE, scale = TRUE)
# Combina todos os ambientes
Geno.all <- rbind(Geno.a, Geno.b, Geno.c, Geno.d)
dim(Geno.all)[1] 982 41456# Calcula matriz de relacionamento genômico
ZG <- tcrossprod(as.matrix(Geno.all)) / ncol(Geno.all)Uma rápida verificação das dimensões e dos primeiros elementos da
matriz ZG para garantir que o cálculo foi bem-sucedido.
# Checagem
dim(ZG)[1] 982 982ZG[1:5, 1:5] A188 A214N A4415 A632 A634
A188 1.081262206 -0.007936875 0.02849732 -0.01341945 0.01086622
A214N -0.007936875 1.106776982 -0.01406327 0.30335381 0.51863619
A4415 0.028497322 -0.014063270 0.97459897 -0.01030808 -0.01225142
A632 -0.013419454 0.303353810 -0.01030808 0.81053742 0.46287155
A634 0.010866216 0.518636190 -0.01225142 0.46287155 1.072240045) Criando a matriz de relacionamento fenômica (ZP)
Seguimos uma lógica idêntica à da seção anterior, mas desta vez usamos os dados espectrais do NIR (que chamamos de ‘P’ de phenomic ou proximal sensing).
- Preparar
NIR: Para cada ambiente (a, b, c, d), removemos as 5 primeiras colunas de metadados (IDs, traits) e usamosscale()para padronizar os valores de cada banda espectral. - Empilhar (
rbind): Combinamos as quatro matrizes NIR escalonadas emNIR.all. - Calcular o Kernel (
tcrossprod): CalculamosZP.
O resultado, ZP, é uma matriz (N total x N total) que
mede a similaridade espectral entre cada par de
observações. A hipótese é que indivíduos com espectros NIR similares
podem compartilhar características fisiológicas ou de composição,
independentemente de seu parentesco genômico.
# Remove colunas de identificação
NIR.a <- scale(a[,-c(1:5)],center=TRUE,scale=TRUE)
NIR.b <- scale(b[,-c(1:5)],center=TRUE,scale=TRUE)
NIR.c <- scale(c[,-c(1:5)],center=TRUE,scale=TRUE)
NIR.d <- scale(d[,-c(1:5)],center=TRUE,scale=TRUE)
# Combina todos os ambientes
NIR.all <- rbind(NIR.a, NIR.b, NIR.c, NIR.d)
dim(NIR.all)[1] 982 3112# Calcula matriz de relacionamento fenômico
ZP <- tcrossprod(as.matrix(NIR.all)) / ncol(NIR.all)Novamente, verificamos as dimensões e o conteúdo da matriz
ZP.
# Checagem
dim(ZP)[1] 982 982ZP[1:5, 1:5] A188 A214N A4415 A632 A634
A188 2.0689756 0.12794826 -0.96853101 1.20893618 0.65051847
A214N 0.1279483 0.01220063 -0.05604141 0.06624954 0.03393302
A4415 -0.9685310 -0.05604141 0.46231635 -0.56784147 -0.29814444
A632 1.2089362 0.06624954 -0.56784147 0.82350392 0.52022985
A634 0.6505185 0.03393302 -0.29814444 0.52022985 0.382097396) Criando a matriz de relacionamento ambiental (ZE)
Esta é a forma mais simples de criar um kernel ambiental.
- Matriz de Incidência (
ZE):model.matrixcria uma matriz “dummy” (ou one-hot encoding). Ela tem uma linha para cada observação e uma coluna para cada ambiente (4 colunas no total). Um valor ‘1’ é colocado na coluna correspondente ao ambiente daquela observação. - Kernel (
ZEZE): Ao calculartcrossprod(ZE), criamos uma matriz (N total x N total) onde o elemento[i, j]é ‘1’ se as observações i e j são do mesmo ambiente, e ‘0’ caso contrário.
Este kernel modela um efeito ambiental “clássico” (ou de bloco), onde todas as observações dentro de um mesmo ambiente são tratadas como 100% relacionadas entre si (em termos de ambiente) e 0% relacionadas a observações de outros ambientes.
# Cria matriz de incidência de ambientes (0/1)
ZE <- model.matrix(~ as.factor(Pheno$Env) - 1)
# Calcula matriz de relacionamento ambiental (produto cruzado)
ZEZE <- tcrossprod(ZE)
# Checagens
cat("Dimensões da matriz ZE:", dim(ZE), "\n")Dimensões da matriz ZE: 982 4 cat("Dimensões da matriz ZEZE:", dim(ZEZE), "\n")Dimensões da matriz ZEZE: 982 982 # Visualiza primeiras linhas
dim(ZE)[1] 982 4ZEZE[1:5, 1:5] 1 2 3 4 5
1 1 1 1 1 1
2 1 1 1 1 1
3 1 1 1 1 1
4 1 1 1 1 1
5 1 1 1 1 16.1) Criando a Matriz de Relacionamento Ambiental com Dados Climáticos (W)
Esta é uma abordagem muito mais sofisticada para
modelar o ambiente, em vez de apenas usar os blocos da Seção 6. O
objetivo é criar um kernel ZW (W de Weather,
Clima) onde a similaridade entre duas observações é baseada na
similaridade climática real dos seus ambientes.
A construção desta matriz W é complexa:
- Carregar ECs: Carregamos as Covariáveis Ambientais (ECs) do script anterior.
- Separar Dados: O código inteligentemente separa os
dados climáticos em dois tipos:
- Agregados: Dados que são um único valor
para o ano (ex:
TMAX_AVG,GDD_CUM).ECs.2011eECs.2012armazenam esses vetores. - Diários: Dados que têm um valor por dia (ex:
PRECTOTCORR).
- Agregados: Dados que são um único valor
para o ano (ex:
- Criar “Assinatura Climática”: O
pivot_wideré usado para transformar os dados diários de um formato “longo” (muitas linhas) para “largo”. O resultado (env_wide_2011) é uma única linha (um vetor) que contém a “assinatura climática” completa daquele ano (ex: T2M_dia1, T2M_dia2, …, T2M_dia150). - Construir Matriz por Observação: Para cada
observação (linha) em um ambiente (ex: ambiente ‘a’, que é 2011):
- Anexamos a “assinatura diária” de 2011 (replicada para aquela linha).
- Anexamos as colunas para os dados diários de 2012, preenchidas com
NA. - Anexamos o vetor de dados “agregados” de 2011.
- Isso é repetido para todos os ambientes (a, b, c, d), preenchendo
com
NAos anos que não se aplicam.
- Empilhar e Limpar:
rbindcombina tudo na matrizW.W[is.na(W)] <- 0substitui osNAs por 0. Isso assume que a “não aplicabilidade” (ex: dados de 2012 para uma observação de 2011) contribui com 0 para a similaridade. - Calcular Kernel
ZW: Finalmente,tcrossprod(W)cria o kernel.ZWé uma matriz (N total x N total) que mede a similaridade climática detalhada entre cada par de observações, usando tanto dados diários quanto agregados.
# --- 1. Preparação Inicial dos Dados Climáticos (Feita uma vez) ---
# Carrega os dados climáticos
ECs_data <- read.csv("output/climate_results/environmental_covariates.csv") %>%
dplyr::select(PRECTOTCORR:date, T_eff_min:VPD_STRESS_DAYS)
# --- 2. Escalonamento dos Dados Climáticos (Feito 1x) ---
# Seleciona apenas as covariáveis AGREGADAS (médias, somas)
ECs_data_scaled <- ECs_data %>%
dplyr::select(TMAX_AVG:VPD_STRESS_DAYS) %>%
scale(., center = TRUE, scale = TRUE) %>% unique()
# Cria ECs.2011 (1 linha com dados agregados e escalonados de 2011)
ECs.2011 <- matrix(ECs_data_scaled[1, ], nrow = 1)
colnames(ECs.2011) <- colnames(ECs_data_scaled)
# Cria ECs.2012 (1 linha com dados agregados e escalonados de 2012)
ECs.2012 <- matrix(ECs_data_scaled[2, ], nrow = 1)
colnames(ECs.2012) <- colnames(ECs_data_scaled)
# Prepara 2011: Filtra dados DIÁRIOS, extrai datas e escala
ECs.2011_filtered <- ECs_data %>% filter(year == 2011)
ECs.2011_scaled <- ECs.2011_filtered %>%
dplyr::select(-year, -date, -(TMAX_AVG:VPD_STRESS_DAYS)) %>%
scale(. , center = TRUE, scale = TRUE)
ECs.2011_dates <- ECs.2011_filtered$date
# Prepara 2012: Filtra dados DIÁRIOS, extrai datas e escala
ECs.2012_filtered <- ECs_data %>% filter(year == 2012)
ECs.2012_scaled <- ECs.2012_filtered %>%
dplyr::select(-year, -date, -(TMAX_AVG:VPD_STRESS_DAYS)) %>%
scale(., center = TRUE, scale = TRUE)
ECs.2012_dates <- ECs.2012_filtered$date
# --- 3. Criar a Linha Única de Dados Climáticos "Largos" (Feito 2x) ---
# Cria o dataframe "largo" (1 linha) para 2011 (assinatura diária)
env_wide_2011 <- as.data.frame(ECs.2011_scaled) %>%
mutate(date = ECs.2011_dates) %>%
pivot_wider(names_from = date, values_from = -date)
# Cria o dataframe "largo" (1 linha) para 2012 (assinatura diária)
env_wide_2012 <- as.data.frame(ECs.2012_scaled) %>%
mutate(date = ECs.2012_dates) %>%
pivot_wider(names_from = date, values_from = -date)
# --- 4. Construção das Matrizes por Ambiente (em etapas) ---
# (Assumindo que os dataframes 'a', 'b', 'c', e 'd' já existem)
# Etapa A: Ambiente CS11_WS
pedigrees_a <- a$Pedigree
ECs.a <- env_wide_2011[rep(1, length(pedigrees_a)), ] # Repete a assinatura diária de 2011
ECs.a <- cbind(ECs.a, matrix(
NA,
nrow = length(pedigrees_a),
ncol = length(env_wide_2012),
dimnames = list(pedigrees_a, colnames(env_wide_2012))
)) # Adiciona colunas NA para 2012
ECs.a <- cbind(ECs.a, ECs.2011) %>% as.matrix() # Adiciona os dados agregados de 2011
rownames(ECs.a) <- pedigrees_a
dim(ECs.a)[1] 196 4607# Etapa B: Ambiente CS11_WW
pedigrees_b <- b$Pedigree
ECs.b <- env_wide_2011[rep(1, length(pedigrees_b)), ]
ECs.b <- cbind(ECs.b, matrix(
NA,
nrow = length(pedigrees_b),
ncol = length(env_wide_2012),
dimnames = list(pedigrees_b, colnames(env_wide_2012))
)) # Adiciona dados de 2012
ECs.b <- cbind(ECs.b, ECs.2011) %>% as.matrix()
rownames(ECs.b) <- pedigrees_b
dim(ECs.b)[1] 263 4607# Etapa C: Ambiente CS12_WS
pedigrees_c <- c$Pedigree
ECs.c <- env_wide_2012[rep(1, length(pedigrees_c)), ] # Repete a assinatura diária de 2012
ECs.c <- cbind(matrix(
NA,
nrow = length(pedigrees_c),
ncol = length(env_wide_2011),
dimnames = list(pedigrees_c, colnames(env_wide_2011))
), ECs.c) # Adiciona colunas NA para 2011
ECs.c <- cbind(ECs.c, ECs.2012) %>% as.matrix() # Adiciona os dados agregados de 2012
rownames(ECs.c) <- pedigrees_c
dim(ECs.c)[1] 310 4607# Etapa D: Ambiente CS12_WW
pedigrees_d <- d$Pedigree
ECs.d <- env_wide_2012[rep(1, length(pedigrees_d)), ]
ECs.d <- cbind(matrix(
NA,
nrow = length(pedigrees_d),
ncol = length(env_wide_2011),
dimnames = list(pedigrees_d, colnames(env_wide_2011))
), ECs.d) # Adiciona dados de 2011
ECs.d <- cbind(ECs.d, ECs.2012) %>% as.matrix()
rownames(ECs.d) <- pedigrees_d
dim(ECs.d)[1] 213 4607# --- 5. Combinação e Criação da Matriz W ---
W <- rbind(ECs.a, ECs.b, ECs.c, ECs.d)
# Verificação
dim(W)[1] 982 4607W[1:5, 1:5] PRECTOTCORR_2011-05-01 PRECTOTCORR_2011-05-02 PRECTOTCORR_2011-05-03
A188 -0.2836821 -0.02845919 -0.2475899
A214N -0.2836821 -0.02845919 -0.2475899
A4415 -0.2836821 -0.02845919 -0.2475899
A632 -0.2836821 -0.02845919 -0.2475899
A634 -0.2836821 -0.02845919 -0.2475899
PRECTOTCORR_2011-05-04 PRECTOTCORR_2011-05-05
A188 -0.2836821 -0.2811041
A214N -0.2836821 -0.2811041
A4415 -0.2836821 -0.2811041
A632 -0.2836821 -0.2811041
A634 -0.2836821 -0.2811041# Substitui NAs por 0 para permitir o cálculo do tcrossprod
W[is.na(W)] <- 0
# 6. Calcular a matriz de relacionamento
ZW <- tcrossprod(as.matrix(W)) / ncol(W)
table(ZW[1,])
-0.00367797795625015 0.498577044593978
523 459 # Visualização do Kernel Ambiental
dim(ZW)[1] 982 982ZW[1:5, 1:5] A188 A214N A4415 A632 A634
A188 0.498577 0.498577 0.498577 0.498577 0.498577
A214N 0.498577 0.498577 0.498577 0.498577 0.498577
A4415 0.498577 0.498577 0.498577 0.498577 0.498577
A632 0.498577 0.498577 0.498577 0.498577 0.498577
A634 0.498577 0.498577 0.498577 0.498577 0.4985777) Verificações Finais de Alinhamento
Antes de prosseguir para a modelagem, executamos algumas verificações cruciais.
Primeiro, uma verificação pontual (spot-check) para um genótipo específico (‘A188’) para ver se os dados parecem corretos e alinhados nas matrizes empilhadas.
Geno.all[str_detect(rownames(Geno.all), "A188"), 1:5] S1_517642 S1_1000282 S1_1763292 S1_1763397 S1_2088440
A188 -0.3244447 -0.4954853 0.4389794 -0.5765720 -0.2468038
A188 -0.3071575 -0.5046434 0.4309819 -0.5753945 -0.2647288
A188 -0.3068291 -0.4844154 0.4318786 -0.5471143 -0.2895119
A188 -0.3277855 -0.4725198 0.4256480 -0.5603790 -0.2964903W[str_detect(rownames(W), "A188"), 1:5] PRECTOTCORR_2011-05-01 PRECTOTCORR_2011-05-02 PRECTOTCORR_2011-05-03
A188 -0.2836821 -0.02845919 -0.2475899
A188 -0.2836821 -0.02845919 -0.2475899
A188 0.0000000 0.00000000 0.0000000
A188 0.0000000 0.00000000 0.0000000
PRECTOTCORR_2011-05-04 PRECTOTCORR_2011-05-05
A188 -0.2836821 -0.2811041
A188 -0.2836821 -0.2811041
A188 0.0000000 0.0000000
A188 0.0000000 0.0000000NIR.all[str_detect(rownames(W), "A188"), 1:5] X3999.640137 X4001.568604 X4003.497071 X4005.425537 X4007.354004
A188 -1.701604 -1.696844 -1.696844 -1.696283 -1.696195
A188 -1.120197 -1.112012 -1.112012 -1.110283 -1.111711
A188 -1.398754 -1.393058 -1.393058 -1.392220 -1.392757
A188 -1.523355 -1.518600 -1.518600 -1.517747 -1.517932Esta é a verificação de alinhamento mais importante
da análise. Antes de passar os kernels para o BGLR, devemos
garantir que todos eles (ZW, ZG,
ZP) são:
- Quadrados (mesmo número de linhas e colunas).
- Simétricos (nomes de linhas == nomes de colunas).
- Perfeitamente alinhados entre si (nomes de linhas de ZW == nomes de linhas de ZG, etc.).
Se todos os testes abaixo retornarem TRUE, nossos
kernels estão perfeitamente alinhados e prontos para a
modelagem.
all(rownames(ZW) == colnames(ZW))[1] TRUEall(rownames(ZG) == colnames(ZG))[1] TRUEall(rownames(ZP) == colnames(ZP))[1] TRUEall(rownames(ZW) == rownames(ZG))[1] TRUEall(rownames(ZW) == rownames(ZP))[1] TRUEall(colnames(ZW) == colnames(ZG))[1] TRUEall(colnames(ZW) == colnames(ZP))[1] TRUEall(unique(rownames(ZG)) == unique(colnames(ZG)))[1] TRUEall(unique(rownames(ZP)) == unique(colnames(ZP)))[1] TRUEall(unique(rownames(ZW)) == unique(colnames(ZW)))[1] TRUEPor fim, inspecionamos os cantos superiores esquerdos das três
matrizes de relacionamento. Isso nos dá uma ideia visual dos valores de
similaridade calculados e confirma que os rownames e
colnames estão idênticos.
ZW[1:5, 1:5] A188 A214N A4415 A632 A634
A188 0.498577 0.498577 0.498577 0.498577 0.498577
A214N 0.498577 0.498577 0.498577 0.498577 0.498577
A4415 0.498577 0.498577 0.498577 0.498577 0.498577
A632 0.498577 0.498577 0.498577 0.498577 0.498577
A634 0.498577 0.498577 0.498577 0.498577 0.498577ZG[1:5, 1:5] A188 A214N A4415 A632 A634
A188 1.081262206 -0.007936875 0.02849732 -0.01341945 0.01086622
A214N -0.007936875 1.106776982 -0.01406327 0.30335381 0.51863619
A4415 0.028497322 -0.014063270 0.97459897 -0.01030808 -0.01225142
A632 -0.013419454 0.303353810 -0.01030808 0.81053742 0.46287155
A634 0.010866216 0.518636190 -0.01225142 0.46287155 1.07224004ZP[1:5, 1:5] A188 A214N A4415 A632 A634
A188 2.0689756 0.12794826 -0.96853101 1.20893618 0.65051847
A214N 0.1279483 0.01220063 -0.05604141 0.06624954 0.03393302
A4415 -0.9685310 -0.05604141 0.46231635 -0.56784147 -0.29814444
A632 1.2089362 0.06624954 -0.56784147 0.82350392 0.52022985
A634 0.6505185 0.03393302 -0.29814444 0.52022985 0.382097398) Interações
Nesta seção, começamos a modelar as interações, como Genótipo-por-Ambiente (GxE) e Fenótipo-por-Ambiente (PxE). Estas interações são cruciais para entender como o desempenho de um genótipo (ou seu perfil NIR) muda em diferentes condições.
O método usado aqui é o produto de Hadamard (a
multiplicação simples, elemento a elemento, * no R). A
lógica é a seguinte:
ZGZE = ZG * ZEZE: Esta matriz representa a interação Genômico × Ambiente (Categórico). Um elemento[i, j]nesta matriz só será diferente de zero se as observações i e j forem do mesmo ambiente (deZEZE) E se elas estiverem genomicamente relacionadas (deZG). Isso isola a variação genômica que é específica de cada ambiente.ZPZE: Da mesma forma, representa a interação Fenômico/NIR × Ambiente (Categórico).ZGZWeZPZW: Representam as interações Genômico × Clima e Fenômico/NIR × Clima. Esta é uma abordagem mais avançada, pois a força da interação não se baseia apenas em “estar no mesmo ambiente” (comoZEZE), mas na “similaridade climática real” (deZW).
# Interação genômica × ambiente
ZGZE <- ZG * ZEZE
ZGZE[1:5, 1:5] A188 A214N A4415 A632 A634
A188 1.081262206 -0.007936875 0.02849732 -0.01341945 0.01086622
A214N -0.007936875 1.106776982 -0.01406327 0.30335381 0.51863619
A4415 0.028497322 -0.014063270 0.97459897 -0.01030808 -0.01225142
A632 -0.013419454 0.303353810 -0.01030808 0.81053742 0.46287155
A634 0.010866216 0.518636190 -0.01225142 0.46287155 1.07224004# Interação fenômica × ambiente
ZPZE <- ZP * ZEZE
ZPZE[1:5, 1:5] A188 A214N A4415 A632 A634
A188 2.0689756 0.12794826 -0.96853101 1.20893618 0.65051847
A214N 0.1279483 0.01220063 -0.05604141 0.06624954 0.03393302
A4415 -0.9685310 -0.05604141 0.46231635 -0.56784147 -0.29814444
A632 1.2089362 0.06624954 -0.56784147 0.82350392 0.52022985
A634 0.6505185 0.03393302 -0.29814444 0.52022985 0.38209739# Interação genômica × clima
ZGZW <- ZG * ZW
ZGZW[1:5, 1:5] A188 A214N A4415 A632 A634
A188 0.539092515 -0.003957144 0.014208110 -0.006690632 0.005417646
A214N -0.003957144 0.551813597 -0.007011624 0.151245246 0.258580099
A4415 0.014208110 -0.007011624 0.485912677 -0.005139370 -0.006108278
A632 -0.006690632 0.151245246 -0.005139370 0.404115351 0.230777129
A634 0.005417646 0.258580099 -0.006108278 0.230777129 0.534594272# Interação fenômica × clima
ZPZW <- ZP * ZW
ZPZW[1:5, 1:5] A188 A214N A4415 A632 A634
A188 1.03154375 0.063792065 -0.48288733 0.6027478 0.32433358
A214N 0.06379207 0.006082952 -0.02794096 0.0330305 0.01691823
A4415 -0.48288733 -0.027940960 0.23050032 -0.2831127 -0.14864797
A632 0.60274783 0.033030500 -0.28311272 0.4105802 0.25937466
A634 0.32433358 0.016918227 -0.14864797 0.2593747 0.190504999) GK and AK kernels
Objetivo geral: Os kernels lineares que
criamos até agora (ZG, ZP) capturam apenas
relações aditivas. No entanto, as interações biológicas
são frequentemente não-lineares. Para capturar esses
padrões complexos, construiremos dois tipos de kernels
não-lineares. Estes kernels serão então usados para construir
os modelos na seção 10.
Tipos de kernels:
- GK (Gaussian Kernel): Mede a similaridade usando uma função gaussiana da distância. É poderoso para capturar “relações locais”, onde indivíduos muito similares têm uma relação muito mais forte do que indivíduos moderadamente similares.
- AK (arc-cosseno Kernel): Mede a similaridade com base no ângulo (correlação) entre os vetores dos indivíduos. É derivado da teoria de redes neurais e é eficaz na captura de padrões complexos e não-aditivos.
9.1) Gaussian kernel (GK)
Construir kernels baseados em funções de similaridade gaussiana, que capturam relações não lineares entre genótipos e fenótipos.
9.1.1) Função de Otimização do Kernel Gaussiano
Antes de construir o kernel, precisamos encontrar o
hiperparâmetro de suavidade (h) ideal. Um
h errado pode fazer o modelo ter overfitting ou
underfitting.
Esta função, margh.fun, é uma “função objetivo” que será
usada por um otimizador (optim). Seu objetivo é encontrar o
valor de h (e um parâmetro de escala phi) que
maximiza a log-verossimilhança marginal.
Em termos simples: dado o nosso kernel candidato
(Kh) e nossos dados fenotípicos (y), a função
calcula o quão “prováveis” são os dados. O otimizador encontrará o
h que torna os dados o mais provável possível.
Kh <- exp(-h * D / q): Constrói o kernel gaussiano candidato.eigenKh: Realiza a decomposição espectral, um passo matemático necessário.lden <- ...: Calcula a log-verossimilhança marginal.return(-(lden + lprior)): Retorna o negativo da log-verossimilhança (pois os otimizadores, por padrão, minimizam).
# Função de otimização do hiperparâmetro h
margh.fun <- function(theta, y, D, q, nu = 0.0001, Sc = 0.0001,
nuh = NULL, Sch = NULL, prior = NULL) {
h <- theta[1] # Parâmetro de suavidade do kernel
phi <- theta[2] # Parâmetro de escala
Kh <- exp(-h * D / q) # Matriz kernel gaussiana
eigenKh <- eigen(Kh) # Decomposição espectral
nr <- length(which(eigenKh$values > 1e-10)) # Número de componentes relevantes
Uh <- eigenKh$vectors[, 1:nr] # Autovetores
Sh <- eigenKh$values[1:nr] # Autovalores
d <- t(Uh) %*% scale(y, scale = FALSE) # Projeção dos dados
# Log-verossimilhança marginal
lden <- -0.5 * sum(log(1 + phi * Sh)) -
(nu + nr - 1) / 2 * log(Sc + sum(d^2 / (1 + phi * Sh)))
# Prior opcional sobre h
if (!is.null(prior)) {
lprior <- dgamma(h, nuh, Sch, log = TRUE)
} else {
lprior <- 0
}
return(-(lden + lprior)) # Retorna negativo da log-verossimilhança
}9.1.2) Construindo o Kernel Gaussiano Genômico (GGK)
Aqui, aplicamos a função de otimização aos nossos dados genômicos.
DG <- (as.matrix(dist(Geno)))^2: Calcula a matriz de distância Euclidiana ao quadrado entre todos os genótipos únicos (ex: 300x300).DG_proj <- ...: Este é um passo crucial.DGé uma matriz de genótipos únicos, mas nosso modelo precisa de um kernel no espaço das observações (ex: 1200x1200). Este código “projeta” a matriz de distânciaDGpara o espaço de observaçãoPhenousando a matriz de incidênciaZ. O resultadoDG_proj[i, j]conterá a distância genômica entre a observação i e a observação j.solG <- optim(...): Executa o otimizador (optim) usandomargh.funpara encontrar o melhorh(baseado no traitPheno$GY).hG <- solG$par[1]: Extrai o valorhotimizado.GGK <- ...: Constrói a matriz kernelGGKfinal usando ohGotimizado.
# Distâncias genômicas
DG <- (as.matrix(dist(Geno)))^2
q05G <- quantile(DG, 0.05)
# Projeta DG para o espaço dos genótipos
IDs <- as.character(Pheno$Pedigree)
DG <- DG[rownames(DG) %in% IDs, rownames(DG) %in% IDs]
IDs <- factor(IDs, levels = rownames(DG))
Z <- as.matrix(model.matrix(~ IDs - 1))
ZD <- tcrossprod(Z, DG)
DG_proj <- tcrossprod(ZD, Z)# Otimização
solG <- optim(c(1, 1), margh.fun, y = Pheno$GY, D = DG_proj, q = q05G,
method = "L-BFGS-B", lower = c(0.05, 0.05), upper = c(6, 30))hG <- solG$par[1]
# Kernel genômico
GGK <- exp(-hG * DG_proj / median(DG_proj))# Checagem
dim(GGK)[1] 982 982GGK[1:5,1:5] 1 2 3 4 5
1 1.0000000 0.8497877 0.8631009 0.8646340 0.8533364
2 0.8497877 1.0000000 0.8538890 0.9055212 0.9197739
3 0.8631009 0.8538890 1.0000000 0.8717643 0.8563903
4 0.8646340 0.9055212 0.8717643 1.0000000 0.9283440
5 0.8533364 0.9197739 0.8563903 0.9283440 1.00000009.1.3) Construindo o Kernel Gaussiano Fenômico (PGK)
Repetimos a mesma lógica, mas desta vez usando os dados espectrais (NIR).
DP <- (as.matrix(dist(NIR.all)))^2: Calcula a matriz de distância Euclidiana ao quadrado diretamente dos dados espectrais (NIR.all). ComoNIR.alljá está no nível de observação (N total x N total), esta matrizDPjá tem a dimensão correta.DP_proj <- ...: Este passo projeta a matrizDP(apesar de já ter a dimensão correta), garantindo que ela esteja perfeitamente alinhada com osrownamesdas outras matrizes kernel.solP <- optim(...): Executa o otimizador para encontrar ohPideal para os dados NIR.PGK <- ...: Constrói o kernelPGKfinal usando ohPotimizado.
# Distâncias fenômicas
DP <- (as.matrix(dist(NIR.all)))^2
q05P <- quantile(DP, 0.05)
IDs <- rownames(NIR.all)
DP <- DP[rownames(DP) %in% IDs, rownames(DP) %in% IDs]
IDs <- IDs
Z <- as.matrix(model.matrix(~ IDs - 1))
Z0 <- tcrossprod(Z, Z)
ZD <- tcrossprod(Z0, DP)
DP_proj <- tcrossprod(ZD, Z0)# Otimização
solP <- optim(c(1, 1), margh.fun, y = Pheno$GY, D = DP_proj, q = q05P,
method = "L-BFGS-B", lower = c(0.05, 0.05), upper = c(6, 30))hP <- solP$par[1]
# Kernel fenômico
PGK <- exp(-hP * DP_proj / median(DP_proj))# Checagem
dim(PGK)[1] 982 982PGK[1:5,1:5] 1 2 3 4 5
1 0.87403262 0.3796616 0.03628784 0.65759840 0.5252460
2 0.37966157 0.9587605 0.55512924 0.30646878 0.7413911
3 0.03628784 0.5551292 0.98313980 0.03056525 0.2830190
4 0.65759840 0.3064688 0.03056525 0.81623029 0.6457647
5 0.52524603 0.7413911 0.28301896 0.64576475 0.98996819.1.4) Interações com Kernels Gaussianos
Agora, podemos criar kernels de interação não-linear usando
o produto de Hadamard com nossos novos kernels GGK
e PGK. Isso nos permite modelar interações GxE e PxE
não-lineares.
- GGKE: interação genômico gaussiano × ambiente (categórico).
- PGKE: interação fenômico gaussiano × ambiente (categórico).
- GGKW: interação genômico gaussiano × clima.
- PGKW: interação fenômico gaussiano × clima.
GGKE <- GGK * ZEZE
PGKE <- PGK * ZEZE
GGKW <- GGK * ZW
PGKW <- PGK * ZW9.2) Arc-cossin Kernels (AK)
Este é um método alternativo para capturar não-linearidade. O kernel Arc-cosseno é baseado no ângulo (ou correlação) entre os vetores de características dos indivíduos.
Uma característica chave deste método é o “nível de recursão”
(nl). Um nl = 1 é como um modelo linear, mas
nl > 1 é análogo a adicionar camadas a uma rede neural,
permitindo que o kernel capture padrões cada vez mais
complexos.
9.2.1) Funções Auxiliares do Kernel Arc-Cosseno
Precisamos de um conjunto de funções para construir e otimizar este kernel.
AK1.fun(X): Constrói o kernel de nível 1 (base). Ele calcula a correlação, converte em ângulo (acos) e então calcula o kernel com base nesse ângulo.AK.fun(AK1, nl): A função de recursão. Ela pega um kernel (comoAK1) e aplica a transformação de arc-cossenonlvezes.marg.AK(y, AK1, ml): A função de otimização. Assim comomargh.fun, ela usa a log-verossimilhança marginal. Ela testa iterativamentenl = 1,nl = 2, etc., até um máximoml, e retorna o nívelnlque maximizou a verossimilhança.
###############################################################
# Código para AK (arc-cosseno Kernel)
# Referência: Crossa et al., 2019
# Objetivo: Ajustar um modelo de predição genômica usando kernel arc-cosseno
# com seleção automática do nível de recursão e modelagem de interação G×E.
###############################################################
# Função para calcular o kernel arc-cosseno de nível 1
AK1.fun <- function(X) {
n <- nrow(X)
cosalfa <- cor(t(X)) # Correlação entre vetores
cosalfa[cosalfa > 1] <- 1
cosalfa[cosalfa < -1] <- -1
angulo <- acos(cosalfa) # Ângulo entre vetores
mag <- sqrt(apply(X, 1, function(x) crossprod(x))) # Magnitude dos vetores
sxy <- tcrossprod(mag) # Produto cruzado das magnitudes
AK1 <- (1 / pi) * sxy * (sin(angulo) + (pi - angulo) * cosalfa)
AK1 <- AK1 / median(AK1, na.rm = TRUE) # Normalização
colnames(AK1) <- rownames(X)
rownames(AK1) <- rownames(X)
return(AK1)
}
# Função para selecionar o nível ótimo de recursão
marg.AK <- function(y, AK1, ml) {
lden.fun <- function(phi, nr, Uh, Sh, d) {
lden <- -0.5 * sum(log((1 + phi * Sh))) - (nr - 1) / 2 * log(sum(d^2 / ((1 + phi * Sh))))
return(-lden)
}
vero <- function(y, GC) {
Kh <- GC
eigenKh <- eigen(Kh)
nr <- length(which(eigenKh$values > 1e-10))
Uh <- eigenKh$vectors[, 1:nr]
Sh <- eigenKh$values[1:nr]
d <- t(Uh) %*% scale(y, scale = FALSE)
sol <- optimize(lden.fun, nr = nr, Uh = Uh, Sh = Sh, d = d,
lower = 0.0005, upper = 200)
phi <- sol[[1]]
log.vero <- 0.5 * sum(log((1 + phi * Sh))) -
(nr - 1) / 2 * log(sum(d^2 / ((1 + phi * Sh))))
return(log.vero)
}
GC <- AK1
l <- 1
GC2 <- GC
vero1 <- vero(y = y, GC = GC2)
m <- 0
while (m == 0 && (l < ml)) {
l <- l + 1
GC <- AK.fun(AK1 = GC2, nl = 1)
GC2 <- GC
vero2 <- vero(y = y, GC = GC2)
if (vero2 < vero1) m = 1
vero1 <- vero2
}
return(l - 1) # Retorna o nível ótimo de recursão
}
# Função para aplicar recursão ao kernel arc-cosseno
AK.fun <- function(AK1, nl) {
n <- nrow(AK1)
AK <- AK1
for (l in 1:nl) {
Aux <- tcrossprod(diag(AK))
cosalfa <- AK * (Aux^(-1 / 2))
cosa <- as.vector(cosalfa)
cosa[cosa > 1] <- 1
cosa[cosa < -1] <- -1
angulo <- acos(cosa)
angulo <- matrix(angulo, n, n)
AK <- (1 / pi) * (Aux^(1 / 2)) * (sin(angulo) + (pi - angulo) * cos(angulo))
}
AK <- AK / median(AK, na.rm = TRUE)
rownames(AK) <- rownames(AK1)
colnames(AK) <- colnames(AK1)
return(AK)
}9.2.2) Construindo o Kernel Arc-Cosseno Genômico (GAK)
GAK1 <- AK1.fun(Geno): Calcula o kernel base (nl=1) a partir dos dados dos genótipos únicos.G <- tcrossprod(...): Projeta o kernelGAK1(ex: 300x300) para o espaço de observação (ex: 1200x1200) para a otimização.Gl <- marg.AK(...): Executa a otimização para encontrar o nível de recursão ótimo (Gl) que melhor explicaPheno$GY.G.L <- tcrossprod(...): Projeta o kernel baseGAK1para o espaço de observação final (similar ao passo 2).GAK <- AK.fun(...): Constrói o kernelGAKfinal aplicando a transformação de arc-cossenoGlvezes.
# Calcula o kernel arc-cosseno de nível 1 com base na matriz de marcadores
GAK1 <- AK1.fun(Geno)
dim(GAK1)[1] 329 329GAK1[1:5, 1:5] A188 A214N A239 A4415 A554
A188 1.7685173 0.9584083 1.0038980 1.0262802 1.0011397
A214N 0.9584083 1.7942427 0.9810621 0.9950431 0.9604347
A239 1.0038980 0.9810621 1.7661590 1.0208122 1.0081672
A4415 1.0262802 0.9950431 1.0208122 1.7922089 1.0261770
A554 1.0011397 0.9604347 1.0081672 1.0261770 1.7531340# Seleciona os IDs do conjunto de dados
IDs <- as.character(Pheno[, "Pedigree"])
GAK1 <- GAK1[rownames(GAK1) %in% IDs, rownames(GAK1) %in% IDs]
# Projeta o kernel para o espaço dos genótipos de treino
IDs <- factor(IDs, levels = rownames(GAK1))
Z <- as.matrix(model.matrix(~ IDs - 1))
G <- tcrossprod(tcrossprod(Z, GAK1), Z)
# Seleciona o nível ótimo de recursão
Gl <- marg.AK(y = Pheno$GY, AK1 = G, ml = 50)
# Projeta o kernel para todos os indivíduos
IDs.L <- Pheno[, "Pedigree"]
IDs.L <- factor(IDs.L, levels = rownames(GAK1))
Z.L <- as.matrix(model.matrix(~ IDs.L - 1))
G.L <- tcrossprod(tcrossprod(Z.L, GAK1), Z.L)
# Aplica recursão
GAK <- AK.fun(AK1 = G.L, nl = Gl)# Checagem
dim(GAK)[1] 982 982GAK[1:5, 1:5] 1 2 3 4 5
1 0.9990646 0.9946313 0.9942027 0.9960061 0.9942696
2 0.9946313 1.0135973 1.0013290 1.0038101 1.0024601
3 0.9942027 1.0013290 1.0124484 1.0027557 1.0009561
4 0.9960061 1.0038101 1.0027557 1.0160907 1.0039067
5 0.9942696 1.0024601 1.0009561 1.0039067 1.01279069.2.3) Construindo o Kernel Arc-Cosseno Fenômico (PAK)
Repetimos a lógica para os dados NIR.
PAK1 <- AK1.fun(NIR.all): Calcula o kernel base (nl=1) a partir dos dados espectrais (NIR.all).P <- tcrossprod(...): Projeta/alinha o kernelPAK1.Pl <- marg.AK(...): Encontra o nível de recursão ótimo (Pl) para os dados NIR.P.L <- tcrossprod(...): Projeta o kernel basePAK1para o espaço de observação final.PAK <- AK.fun(...): Constrói o kernelPAKfinal aplicando a transformaçãoPlvezes.
# Calcula o kernel arc-cosseno de nível 1 com base na matriz fenômica
PAK1 <- AK1.fun(NIR.all)
dim(PAK1)[1] 982 982PAK1[1:5, 1:5] A188 A214N A4415 A632 A634
A188 20.11384819 1.0354311 1.5396601 0.04303532 0.1343575
A214N 1.03543108 0.1186102 0.3223817 0.13156365 0.1268015
A4415 1.53966005 0.3223817 4.4944758 4.08786393 3.2061644
A632 0.04303532 0.1315636 4.0878639 8.00581345 5.3267106
A634 0.13435748 0.1268015 3.2061644 5.32671059 3.7146155# Seleciona os IDs do conjunto de dados
IDs <- as.character(rownames(NIR.all))
PAK1 <- PAK1[rownames(PAK1) %in% IDs, rownames(PAK1) %in% IDs]
# Projeta o kernel para o espaço dos fenótipos
IDs <- IDs
Z <- as.matrix(model.matrix(~ IDs - 1))
Z0 <- tcrossprod(Z, Z)
ZD <- tcrossprod(Z0, PAK1)
P <- tcrossprod(ZD, Z0)
# Seleciona o nível ótimo de recursão
Pl <- marg.AK(y = Pheno$GY, AK1 = P, ml = 50)
# Projeta o kernel para todos os indivíduos
IDs.L <- rownames(NIR.all)
Z.L <- as.matrix(model.matrix(~ IDs.L - 1))
Z0.L <- tcrossprod(Z.L, Z.L)
P.L <- tcrossprod(tcrossprod(Z0.L, PAK1), Z0.L)
# Aplica recursão
PAK <- AK.fun(AK1 = P.L, nl = Pl)dim(PAK)[1] 982 982PAK[1:5, 1:5] 1 2 3 4 5
1 6.8121174 0.8792315 1.3764872 2.3285678 1.0422713
2 0.8792315 0.1710425 0.3189383 0.4146304 0.1960632
3 1.3764872 0.3189383 1.1884678 1.8624650 0.9143113
4 2.3285678 0.4146304 1.8624650 7.7687820 3.3814512
5 1.0422713 0.1960632 0.9143113 3.3814512 1.52295729.2.4) Interações com Arc-cosseno Kernels
Finalmente, criamos os kernels de interação não-linear
finais usando o produto de Hadamard com GAK e
PAK.
- GAKE: interação GAK × Ambiente (Categórico).
- PAKE: interação PAK × Ambiente (Categórico).
- GAKW: interação GAK × Clima (Baseado em ECs).
- PAKW: interação PAK × Clima (Baseado em ECs).
# GAK × Ambiente
GAKE <- GAK * ZEZE
GAKE[1:5,1:5] 1 2 3 4 5
1 0.9990646 0.9946313 0.9942027 0.9960061 0.9942696
2 0.9946313 1.0135973 1.0013290 1.0038101 1.0024601
3 0.9942027 1.0013290 1.0124484 1.0027557 1.0009561
4 0.9960061 1.0038101 1.0027557 1.0160907 1.0039067
5 0.9942696 1.0024601 1.0009561 1.0039067 1.0127906# PAK × Ambiente
PAKE <- PAK * ZEZE
PAKE[1:5,1:5] 1 2 3 4 5
1 6.8121174 0.8792315 1.3764872 2.3285678 1.0422713
2 0.8792315 0.1710425 0.3189383 0.4146304 0.1960632
3 1.3764872 0.3189383 1.1884678 1.8624650 0.9143113
4 2.3285678 0.4146304 1.8624650 7.7687820 3.3814512
5 1.0422713 0.1960632 0.9143113 3.3814512 1.5229572# GAK × Ambiente
GAKW <- GAK * ZW
GAKW[1:5,1:5] 1 2 3 4 5
1 0.4981107 0.4959003 0.4956866 0.4965858 0.4957200
2 0.4959003 0.5053563 0.4992396 0.5004767 0.4998036
3 0.4956866 0.4992396 0.5047835 0.4999510 0.4990537
4 0.4965858 0.5004767 0.4999510 0.5065995 0.5005249
5 0.4957200 0.4998036 0.4990537 0.5005249 0.5049541# PAK × Ambiente
PAKW <- PAK * ZW
PAKW[1:5,1:5] 1 2 3 4 5
1 3.3963654 0.43836464 0.6862849 1.1609705 0.51965253
2 0.4383646 0.08527786 0.1590153 0.2067252 0.09775261
3 0.6862849 0.15901531 0.5925428 0.9285823 0.45585462
4 1.1609705 0.20672521 0.9285823 3.8733364 1.68591397
5 0.5196525 0.09775261 0.4558546 1.6859140 0.7593115210) Limpar o ambiente
Esta é uma etapa crucial de gerenciamento de memória e checkpointing (salvamento de progresso).
O processo de criação de kernels (especialmente os
kernels GK e AK nas seções 8 e 9) é computacionalmente e
intensivo em termos de memória. Ele cria muitos objetos intermediários
grandes (ex: DG, DP, solG,
GAK1, Geno.a, etc.) que não são mais
necessários para a modelagem.
Se não limpássemos o ambiente, correríamos um sério risco de ficar
sem RAM ao tentar ajustar os modelos BGLR na próxima
seção.
A estratégia aqui é dupla:
- Checkpointing (Cache): Primeiro, identificamos
todos os objetos essenciais que precisamos para a próxima etapa
(nossos kernels finais e
Pheno) na listakeep_names. Em seguida, salvamos cada um deles como um arquivo.rdsindividual. Isso atua como um ponto de salvamento. Se o R travar durante a modelagem, podemos reiniciar e carregar esses arquivos diretamente, sem ter que re-executar todo o script (o que poderia levar horas). - Redefinição de Memória (Hard Reset): Após salvar, o
script remove todos os outros objetos (
to_remove) da memória. Em seguida, ele chama o “coletor de lixo” (gc()) e recarrega imediatamente os arquivos.rdsque acabamos de salvar. Este processo de salvar, limpar tudo e recarregar é a maneira mais robusta de garantir que o R libere toda a memória “lixo” e comece a próxima etapa com o ambiente mais limpo possível.
# Liste aqui os objetos que deseja manter no ambiente:
keep_names <- c(
# matrizes / objetos para modelos
"ZE", "ZG", "ZP", "ZGZE", "ZPZE", "ZW", "ZGZW", "ZPZW",
# Kernels
"GGK", "PGK", "GGKE", "PGKE", "GGKW", "PGKW",
"GAK", "PAK", "GAKE", "PAKE", "GAKW", "PAKW",
# outros objetos importantes
"Pheno", "Pedigree"
)
# Confirmação (opcional): mostra o que será removido e o que será mantido
current_objs <- ls(envir = .GlobalEnv)
to_keep <- intersect(current_objs, keep_names)
to_remove <- setdiff(current_objs, to_keep)
message("Objetos que serão mantidos (", length(to_keep), "): ", paste(to_keep, collapse = ", "))
message("Objetos que serão removidos (", length(to_remove), "): ", paste(head(to_remove, 40), collapse = ", "),
if (length(to_remove) > 40) paste0(" ... (+", length(to_remove) - 40, " mais)"))
# Salva cada objeto como .rds individualmente
for (obj_name in to_keep) {
saveRDS(get(obj_name), file = file.path("output/Matrizes", paste0(obj_name, ".rds")))
}
# Executa remoção
rm(list = to_remove, envir = .GlobalEnv)
gc() used (Mb) gc trigger (Mb) max used (Mb)
Ncells 1604970 85.8 2516773 134.5 2516773 134.5
Vcells 21048481 160.6 175727516 1340.7 219659394 1675.9# Lista todos os arquivos .rds na pasta output/Matrizes
rds_files <- list.files("output/Matrizes", pattern = "\\.rds$", full.names = TRUE)
# Carrega cada objeto e atribui ao ambiente global com seu nome original
for (file in rds_files) {
obj_name <- tools::file_path_sans_ext(basename(file))
assign(obj_name, readRDS(file), envir = .GlobalEnv)
}
# Confirmação opcional
message("Objetos carregados: ", paste(tools::file_path_sans_ext(basename(rds_files)), collapse = ", "))11) Modelos preditivos
Esta seção é o “plano de arquitetura” da nossa análise. Tendo passado todo o trabalho de criação de mais de uma dúzia de kernels (lineares, não-lineares e de interação), agora definimos formalmente os modelos que iremos testar.
O objetivo é comparar sistematicamente diferentes hipóteses: * O
kernel NIR (P) é melhor que o genômico (G)? *
Os kernels não-lineares (GGK, GAK)
capturam mais variação do que os lineares? * A interação GxE categórica
(GE) é suficiente, ou a interação baseada no clima
(GW) é melhor? * O que acontece quando combinamos
kernels (ex: G+P)?
11.1) Notação dos Efeitos
A notação padronizada abaixo descreve cada componente (ou “bloco de
construção”) que usaremos nos modelos. Cada um deles é uma matriz
kernel que passaremos para o BGLR.
- E → Efeito principal de Ambiente
(Environment), categórico. (Nosso kernel
ZEZE). - W → Efeito principal do Clima
(Climate), baseado no kernel de covariáveis ambientais. (Nosso
kernel
ZW). - G → Efeito Genômico
(Genomic), kernel linear. (Nosso kernel
ZG). - P → Efeito Fenômico
(Phenomic, ex.: bandas NIRS), kernel linear. (Nosso kernel
ZP). - GGK → Efeito Genômico GK
(Genomic Gaussian Kernel). (Nosso kernel
GGK). - PGK → Efeito Fenômico GK
(Phenomic Gaussian Kernel). (Nosso kernel
PGK). - GAK → Efeito Genômico A-Kernel
(Genomic Arc-Kernel). (Nosso kernel
GAK). - PAK → Efeito Fenômico A-Kernel
(Phenomic Arc-Kernel). (Nosso kernel
PAK).
Interações com Ambiente Categórico (E)
Estes são os kernels de interação que usam o produto de
Hadamard com ZEZE. Eles modelam a variação que é específica
para um bloco ambiental (ex: CS11_WS).
- GE → Interação Genômico × Ambiente
(G×E). (Kernel
ZGZE). - PE → Interação Fenômico × Ambiente
(P×E). (Kernel
ZPZE). - GGKE → Interação Genômico GK ×
Ambiente (GGK×E). (Kernel
GGKE). - PGKE → Interação Fenômico GK ×
Ambiente (PGK×E). (Kernel
PGKE). - GAKE → Interação Genômico A-Kernel ×
Ambiente (GAK×E). (Kernel
GAKE). - PAKE → Interação Fenômico A-Kernel ×
Ambiente (PAK×E). (Kernel
PAKE).
Interações com Ambiente Climático (W)
Estes são os kernels de interação que usam o produto de
Hadamard com ZW. Eles modelam a variação que é específica
para perfis climáticos similares.
- GW → Interação Genômico × Clima
(G×W). (Kernel
ZGZW). - PW → Interação Fenômico × Clima
(P×W). (Kernel
ZPZW). - (Outras interações como GGKW, PGKW, etc., seguem a mesma lógica.)
Estrutura dos Modelos
Os modelos são agrupados por complexidade e pela hipótese que estão
testando. Cada um será definido no BGLR como uma lista de
kernels.
Grupo 1: Modelos Base (Ambiente Categórico E)
Hipótese: “A variação ambiental é melhor explicada
por blocos categóricos (E = ZEZE).”
1.1: Modelos de Efeitos Principais
Objetivo: Testar cada kernel (G, P, GGK, PGK, etc.)
isoladamente para ver qual deles, sozinho, melhor captura a variação não
explicada pelo ambiente E.
| Modelo | Estrutura de Efeitos |
|---|---|
| E+G | Ambiente + Genômico (Linear) |
| E+P | Ambiente + Fenômico (Linear) |
| E+GGK | Ambiente + Genômico (Gaussiano) |
| E+PGK | Ambiente + Fenômico (Gaussiano) |
| E+GAK | Ambiente + Genômico (Arc-Kernel) |
| E+PAK | Ambiente + Fenômico (Arc-Kernel) |
1.2: Modelos com Interação G×E Objetivo:
Testar se adicionar um termo de interação GxE (baseado em
ZEZE) ao seu respectivo efeito principal melhora a
predição.
| Modelo | Estrutura de Efeitos |
|---|---|
| E+G+GE | Ambiente + Genômico + Interação G×E |
| E+P+PE | Ambiente + Fenômico + Interação P×E |
| E+GGK+GGKE | Ambiente + Genômico (Gaussiano) + Interação GGK×E |
| E+PGK+PGKE | Ambiente + Fenômico (Gaussiano) + Interação PGK×E |
| E+GAK+GAKE | Ambiente + Genômico (Arc-Kernel) + Interação GAK×E |
| E+PAK+PAKE | Ambiente + Fenômico (Arc-Kernel) + Interação PAK×E |
1.3: Modelos Combinados (G + P) Objetivo:
Testar se os kernels genômicos e fenômicos (NIR) capturam
fontes de variação diferentes e, portanto, se sua combinação
(G+P) é melhor do que qualquer um deles sozinho.
| Modelo | Estrutura de Efeitos |
|---|---|
| E+G+P | Ambiente + Genômico + Fenômico |
| E+GGK+PGK | Ambiente + Genômico (Gaussiano) + Fenômico (Gaussiano) |
| E+GAK+PAK | Ambiente + Genômico (Arc-Kernel) + Fenômico (Arc-Kernel) |
| E+G+P+GE+PE | Ambiente + Genômico + Fenômico + Ambas Interações G×E |
| E+GGK+PGK+GGKE+PGKE | Ambiente + G (Gaussiano) + P (Gaussiano) + Ambas Interações G×E |
| E+GAK+PAK+GAKE+PAKE | Ambiente + G (Arc-Kernel) + P (Arc-Kernel) + Ambas Interações G×E |
Grupo 2: Modelos com Covariáveis Climáticas (W)
Hipótese: “A variação ambiental é melhor explicada
por dados climáticos reais (W = ZW) do que por blocos
categóricos.”
2.1: Modelos com Efeitos Climáticos Aditivos (E + W)
Objetivo: Testar se adicionar o kernel climático
(W) como um efeito principal adicional (junto com
E) melhora o modelo. Isso permite que o modelo capture
tanto a variação do bloco E quanto a variação explicada
pelo clima W.
| Modelo | Estrutura de Efeitos |
|---|---|
| E+W | Ambiente (Categórico) + Clima (Kernel) |
| E+W+G | E+W + Genômico (Linear) |
| E+W+P | E+W + Fenômico (Linear) |
| E+W+G+P | E+W + Genômico + Fenômico |
| E+W+GGK | E+W + Genômico (Gaussiano) |
| E+W+PGK | E+W + Fenômico (Gaussiano) |
| E+W+GGK+PGK | E+W + G (Gaussiano) + P (Gaussiano) |
| E+W+GAK | E+W + Genômico (Arc-Kernel) |
| E+W+PAK | E+W + Fenômico (Arc-Kernel) |
| E+W+GAK+PAK | E+W + G (Arc-Kernel) + P (Arc-Kernel) |
2.2: Modelos Climáticos com Interações
Objetivo: Testar diferentes formas de interação na presença do
kernel climático W.
Subgrupo 2.2.1: Interação Categórica (G×E) mantida
(Hipótese: “Mesmo com W no modelo, a interação G×E categórica
(GE) ainda captura variação útil que W não
captura.”)
| Modelo | Estrutura de Efeitos |
|---|---|
| E+W+G+GE | E+W + Genômico (Linear) + Interação G×E |
| E+W+P+PE | E+W + Fenômico (Linear) + Interação P×E |
| E+W+G+P+GE+PE | E+W + G + P + Ambas Interações G×E |
| E+W+GGK+GGKE | E+W + G (Gaussiano) + Interação GGK×E |
| … (etc.) | (Modelos equivalentes para PGK, GAK, PAK) |
| E+W+GAK+PAK+GAKE+PAKE | E+W + G/P (Arc-Kernel) + Ambas Interações G×E |
Subgrupo 2.2.2: Interação Climática (G×W) (Hipótese:
“Uma interação baseada no clima real (GW) é uma forma mais
precisa e parcimoniosa de modelar GxE do que a interação categórica
GE.”)
| Modelo | Estrutura de Efeitos |
|---|---|
| E+W+G+GW | E+W + Genômico (Linear) + Interação G×W |
| E+W+P+PW | E+W + Fenômico (Linear) + Interação P×W |
| E+W+G+P+GW+PW | E+W + G + P + Ambas Interações G×W |
| E+W+GGK+GGKW | E+W + G (Gaussiano) + Interação GGK×W |
| … (etc.) | (Modelos equivalentes para PGK, GAK, PAK) |
| E+W+GAK+PAK+GAKW+PAKW | E+W + G/P (Arc-Kernel) + Ambas Interações G×W |
2.3: Modelos Climáticos sem Efeito Categórico (W substitui
E) Objetivo: Testar a hipótese mais forte: “O
kernel climático W captura toda a
variação ambiental relevante, tornando o efeito categórico
E desnecessário.”
| Modelo | Estrutura de Efeitos |
|---|---|
| W | Clima (Kernel) |
| W+G | Clima (Kernel) + Genômico (Linear) |
| W+P | Clima (Kernel) + Fenômico (Linear) |
| W+G+P | Clima (Kernel) + Genômico + Fenômico |
| W+GGK | Clima (Kernel) + Genômico (Gaussiano) |
| W+PGK | Clima (Kernel) + Fenômico (Gaussiano) |
| W+GGK+PGK | Clima (Kernel) + G (Gaussiano) + P (Gaussiano) |
| W+GAK | Clima (Kernel) + Genômico (Arc-Kernel) |
| W+PAK | Clima (Kernel) + Fenômico (Arc-Kernel) |
| W+GAK+PAK | Clima (Kernel) + G (Arc-Kernel) + P (Arc-Kernel) |
11.2) Tabela de classificação dos modelos
Objetivo: Criar um “dicionário” ou “tabela de consulta” para nossos modelos.
Nossos dados de resultados agora têm uma coluna Model
com valores como “M1”, “M2”, … “M62”. Isso não é informativo. Para
nossos gráficos e tabelas finais, queremos poder dizer que “M1” é “E +
G” e que “M17” é “E + GGK + PGK + GGKE + PGKE”.
Este bloco de código cria o dataframe
model_info, que mapeia:
ModelID: O número (1-62)Model: O rótulo (“M1” - “M62”)Type: Uma categoria de alto nível (ex: “Basico”, “Clima_IntW”, “Individual”)Description: O rótulo legível (ex: “E + G + GE”)
Este dataframe será “juntado” (left_join) aos
nossos dados de resultados antes da plotagem.
# --- Cria a tabela de classificação dos modelos (Completa) ---
model_info <- tibble(
ModelID = 1:62,
Model = paste0("M", ModelID),
Type = case_when(
# Grupo 1: Modelos Base (Efeito Categórico E)
ModelID %in% c(1:6, 13:15) ~ "Basico", # E + G/P (efeitos principais)
ModelID %in% c(7:12, 16:18) ~ "Interacao_E", # E + G/P + GxE/PxE
# Grupo 2: Modelos Climáticos (E + W)
ModelID %in% 19:27 ~ "Clima_Base_E", # E + W + G/P (efeitos principais)
ModelID %in% 28:36 ~ "Clima_IntE", # E + W + G/P + GxE/PxE
ModelID %in% 37:45 ~ "Clima_IntW", # E + W + G/P + GxW/PxW
# Grupo 3: Modelos Climáticos (W substitui E)
ModelID %in% 46:54 ~ "Clima_Base_noE", # W + G/P (sem E fixo),
# Grrupo 4: Modelos individuais
ModelID %in% c(55:62) ~ "Individual",
TRUE ~ "Outros"
),
Description = case_when(
# Bloco 1: (Item 10) Modelos básicos (E + G/P)
ModelID == 1 ~ "E + G",
ModelID == 2 ~ "E + P",
ModelID == 3 ~ "E + GGK",
ModelID == 4 ~ "E + PGK", # (Baseado em Eta4)
ModelID == 5 ~ "E + GAK", # (Baseado em Eta5)
ModelID == 6 ~ "E + PAK",
# Bloco 2: (Item 10) Modelos com interações categóricas (E + G/P + GxE/PxE)
ModelID == 7 ~ "E + G + GE",
ModelID == 8 ~ "E + P + PE",
ModelID == 9 ~ "E + GGK + GGKE",
ModelID == 10 ~ "E + PGK + PGKE", # (Baseado em Eta10)
ModelID == 11 ~ "E + GAK + GAKE", # (Baseado em Eta11)
ModelID == 12 ~ "E + PAK + PAKE",
# Bloco 3: (Item 10) Modelos combinados (E + G + P)
ModelID == 13 ~ "E + G + P",
ModelID == 14 ~ "E + GGK + PGK",
ModelID == 15 ~ "E + GAK + PAK",
# Bloco 4: (Item 10) Modelos combinados com interação categórica
ModelID == 16 ~ "E + G + P + GE + PE",
ModelID == 17 ~ "E + GGK + PGK + GGKE + PGKE",
ModelID == 18 ~ "E + GAK + PAK + GAKE + PAKE",
# Bloco 5: (Item 10.1) Modelos E + W + G/P (Clima_Base_E)
ModelID == 19 ~ "E + G + W",
ModelID == 20 ~ "E + P + W",
ModelID == 21 ~ "E + G + P + W",
ModelID == 22 ~ "E + GGK + W",
ModelID == 23 ~ "E + PGK + W",
ModelID == 24 ~ "E + GGK + PGK + W",
ModelID == 25 ~ "E + GAK + W",
ModelID == 26 ~ "E + PAK + W",
ModelID == 27 ~ "E + GAK + PAK + W",
# Bloco 6: (Item 10.1) Modelos E + W + G/P + GxE/PxE (Clima_IntE)
ModelID == 28 ~ "E + G + W + GE",
ModelID == 29 ~ "E + P + W + PE",
ModelID == 30 ~ "E + G + P + W + GE + PE",
ModelID == 31 ~ "E + GGK + W + GGKE",
ModelID == 32 ~ "E + PGK + W + PGKE",
ModelID == 33 ~ "E + GGK + PGK + W + GGKE + PGKE",
ModelID == 34 ~ "E + GAK + W + GAKE",
ModelID == 35 ~ "E + PAK + W + PAKE",
ModelID == 36 ~ "E + GAK + PAK + W + GAKE + PAKE",
# Bloco 7: (Item 10.1) Modelos E + W + G/P + GxW/PxW (Clima_IntW)
ModelID == 37 ~ "E + G + W + GW",
ModelID == 38 ~ "E + P + W + PW",
ModelID == 39 ~ "E + G + P + W + GW + PW",
ModelID == 40 ~ "E + GGK + W + GGKW",
ModelID == 41 ~ "E + PGK + W + PGKW",
ModelID == 42 ~ "E + GGK + PGK + W + GGKW + PGKW",
ModelID == 43 ~ "E + GAK + W + GAKW",
ModelID == 44 ~ "E + PAK + W + PAKW",
ModelID == 45 ~ "E + GAK + PAK + W + GAKW + PAKW",
# Bloco 8: (Item 10.1) Modelos W + G/P (Sem E) (Clima_Base_noE)
ModelID == 46 ~ "G + W",
ModelID == 47 ~ "P + W",
ModelID == 48 ~ "G + P + W",
ModelID == 49 ~ "GGK + W",
ModelID == 50 ~ "PGK + W",
ModelID == 51 ~ "GGK + PGK + W",
ModelID == 52 ~ "GAK + W",
ModelID == 53 ~ "PAK + W",
ModelID == 54 ~ "GAK + PAK + W",
# Bloco 9: (Item 11.5) Modelos individuais
ModelID == 55 ~ "E",
ModelID == 56 ~ "G",
ModelID == 57 ~ "P",
ModelID == 58 ~ "GGK",
ModelID == 59 ~ "PGK",
ModelID == 60 ~ "GAK",
ModelID == 61 ~ "PAK",
ModelID == 62 ~ "W",
TRUE ~ as.character(ModelID) # Fallback
)
)12) Definição dos modelos preditivos
Nesta seção, traduzimos nosso “plano de arquitetura” da Seção 11 em
código. Cada modelo (Eta1, Eta2, etc.) é definido como um objeto
list do R, que é o formato exato que o pacote
BGLR exige para seu argumento ETA (preditor
linear).
A sintaxe do BGLR que estamos usando é:
list(X = ZE, model = "BRR"): UsamosBRR(Bayesian Ridge Regression) para o efeito de ambienteE. Isso trataEcomo um efeito fixo (em um contexto Bayesiano) e passamos a matriz de incidênciaZEcomo o preditorX.list(K = ZG, model = "RKHS"): UsamosRKHS(Reproducing Kernel Hilbert Spaces) para todos os nossos efeitos aleatórios (G, P, GxE, etc.). Isso instrui oBGLRa usar a nossa matriz kernel pré-calculada (ex:ZG) para o argumentoK.
Cada objeto Eta é uma “receita” de modelo que o
BGLR irá ajustar.
# -------------------------------------------------------------------
# GRUPO 1: MODELOS BASE (EFEITO CATEGÓRICO 'E')
# (Modelos Eta1 a Eta18)
# -------------------------------------------------------------------
# --- 1.1: Efeitos Principais (E + G/P) ---
# Modelo 1: E + G (Linear)
Eta1 <- list(
E = list(X = ZE, model = "BRR"),
G = list(K = ZG, model = "RKHS")
)
# Modelo 2: E + P (Linear)
Eta2 <- list(
E = list(X = ZE, model = "BRR"),
P = list(K = ZP, model = "RKHS")
)
# Modelo 3: E + GGK (Gaussiano)
Eta3 <- list(
E = list(X = ZE, model = "BRR"),
GGK = list(K = GGK, model = "RKHS")
)
# Modelo 4: E + PGK (Gaussiano)
Eta4 <- list(
E = list(X = ZE, model = "BRR"),
PGK = list(K = PGK, model = "RKHS")
)
# Modelo 5: E + GAK (Arc-cosseno)
Eta5 <- list(
E = list(X = ZE, model = "BRR"),
GAK = list(K = GAK, model = "RKHS")
)
# Modelo 6: E + PAK (Arc-cosseno)
Eta6 <- list(
E = list(X = ZE, model = "BRR"),
PAK = list(K = PAK, model = "RKHS")
)
# Modelo 13: E + G + P (Linear)
Eta13 <- list(
E = list(X = ZE, model = "BRR"),
G = list(K = ZG, model = "RKHS"),
P = list(K = ZP, model = "RKHS")
)
# Modelo 14: E + GGK + PGK (Gaussiano)
Eta14 <- list(
E = list(X = ZE, model = "BRR"),
GGK = list(K = GGK, model = "RKHS"),
PGK = list(K = PGK, model = "RKHS")
)
# Modelo 15: E + GAK + PAK (Arc-cosseno)
Eta15 <- list(
E = list(X = ZE, model = "BRR"),
GAK = list(K = GAK, model = "RKHS"),
PAK = list(K = PAK, model = "RKHS")
)
# --- 1.2: Efeitos Principais + Interação GxE (E + G/P + GxE/PxE) ---
# Modelo 7: E + G + GE (Linear)
Eta7 <- list(
E = list(X = ZE, model = "BRR"),
G = list(K = ZG, model = "RKHS"),
GE = list(K = ZGZE, model = "RKHS")
)
# Modelo 8: E + P + PE (Linear)
Eta8 <- list(
E = list(X = ZE, model = "BRR"),
P = list(K = ZP, model = "RKHS"),
PE = list(K = ZPZE, model = "RKHS")
)
# Modelo 9: E + GGK + GGKE (Gaussiano)
Eta9 <- list(
E = list(X = ZE, model = "BRR"),
GGK = list(K = GGK, model = "RKHS"),
GGKE = list(K = GGKE, model = "RKHS")
)
# Modelo 10: E + PGK + PGKE (Gaussiano)
Eta10 <- list(
E = list(X = ZE, model = "BRR"),
PGK = list(K = PGK, model = "RKHS"),
PGKE = list(K = PGKE, model = "RKHS")
)
# Modelo 11: E + GAK + GAKE (Arc-cosseno)
Eta11 <- list(
E = list(X = ZE, model = "BRR"),
GAK = list(K = GAK, model = "RKHS"),
GAKE = list(K = GAKE, model = "RKHS")
)
# Modelo 12: E + PAK + PAKE (Arc-cosseno)
Eta12 <- list(
E = list(X = ZE, model = "BRR"),
PAK = list(K = PAK, model = "RKHS"),
PAKE = list(K = PAKE, model = "RKHS")
)
# Modelo 16: E + G + P + GE + PE (Linear)
Eta16 <- list(
E = list(X = ZE, model = "BRR"),
G = list(K = ZG, model = "RKHS"),
P = list(K = ZP, model = "RKHS"),
GE = list(K = ZGZE, model = "RKHS"),
PE = list(K = ZPZE, model = "RKHS")
)
# Modelo 17: E + GGK + PGK + GGKE + PGKE (Gaussiano)
Eta17 <- list(
E = list(X = ZE, model = "BRR"),
GGK = list(K = GGK, model = "RKHS"),
PGK = list(K = PGK, model = "RKHS"),
GGKE = list(K = GGKE, model = "RKHS"),
PGKE = list(K = PGKE, model = "RKHS")
)
# Modelo 18: E + GAK + PAK + GAKE + PAKE (Arc-cosseno)
Eta18 <- list(
E = list(X = ZE, model = "BRR"),
GAK = list(K = GAK, model = "RKHS"),
PAK = list(K = PAK, model = "RKHS"),
GAKE = list(K = GAKE, model = "RKHS"),
PAKE = list(K = PAKE, model = "RKHS")
)
# -------------------------------------------------------------------
# GRUPO 2: MODELOS CLIMÁTICOS (EFEITOS 'E' + 'W')
# (Modelos Eta19 a Eta46)
# -------------------------------------------------------------------
# --- 2.1: Efeitos Principais (E + W + G/P) ---
# Modelo 19: E + W (Linha de base climática)
Eta19 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, model = "RKHS")
)
# Modelo 20: E + W + G (Linear)
Eta20 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, model = "RKHS"),
G = list(K = ZG, model = "RKHS")
)
# Modelo 21: E + W + P (Linear)
Eta21 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, model = "RKHS"),
P = list(K = ZP, model = "RKHS")
)
# Modelo 22: E + W + G + P (Linear)
Eta22 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, model = "RKHS"),
G = list(K = ZG, model = "RKHS"),
P = list(K = ZP, model = "RKHS")
)
# Modelo 23: E + W + GGK (Gaussiano)
Eta23 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, model = "RKHS"),
GGK = list(K = GGK, model = "RKHS")
)
# Modelo 24: E + W + PGK (Gaussiano)
Eta24 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, model = "RKHS"),
PGK = list(K = PGK, model = "RKHS")
)
# Modelo 25: E + W + GGK + PGK (Gaussiano)
Eta25 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, model = "RKHS"),
GGK = list(K = GGK, model = "RKHS"),
PGK = list(K = PGK, model = "RKHS")
)
# Modelo 26: E + W + GAK (Arc-cosseno)
Eta26 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
GAK = list(K = GAK, model = "RKHS")
)
# Modelo 27: E + W + PAK (Arc-cosseno)
Eta27 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
PAK = list(K = PAK, model = "RKHS")
)
# Modelo 28: E + W + GAK + PAK (Arc-cosseno)
Eta28 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
GAK = list(K = GAK, model = "RKHS"),
PAK = list(K = PAK, model = "RKHS")
)
# --- 2.2: Interação com Ambiente Categórico (E + W + G/P + GxE/PxE) ---
# Modelo 29: E + W + G + GE (Linear)
Eta29 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
G = list(K = ZG, model = "RKHS"),
GE = list(K = ZGZE, model = "RKHS")
)
# Modelo 30: E + W + P + PE (Linear)
Eta30 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
P = list(K = ZP, model = "RKHS"),
PE = list(K = ZPZE, model = "RKHS")
)
# Modelo 31: E + W + G + P + GE + PE (Linear)
Eta31 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
G = list(K = ZG, model = "RKHS"),
P = list(K = ZP, model = "RKHS"),
GE = list(K = ZGZE, model = "RKHS"),
PE = list(K = ZPZE, model = "RKHS")
)
# Modelo 32: E + W + GGK + GGKE (Gaussiano)
Eta32 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
GGK = list(K = GGK, model = "RKHS"),
GGKE = list(K = GGKE, model = "RKHS")
)
# Modelo 33: E + W + PGK + PGKE (Gaussiano)
Eta33 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
PGK = list(K = PGK, model = "RKHS"),
PGKE = list(K = PGKE, model = "RKHS")
)
# Modelo 34: E + W + GGK + PGK + GGKE + PGKE (Gaussiano)
Eta34 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
GGK = list(K = GGK, model = "RKHS"),
PGK = list(K = PGK, model = "RKHS"),
GGKE = list(K = GGKE, model = "RKHS"),
PGKE = list(K = PGKE, model = "RKHS")
)
# Modelo 35: E + W + GAK + GAKE (Arc-cosseno)
Eta35 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
GAK = list(K = GAK, model = "RKHS"),
GAKE = list(K = GAKE, model = "RKHS")
)
# Modelo 36: E + W + PAK + PAKE (Arc-cosseno)
Eta36 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
PAK = list(K = PAK, model = "RKHS"),
PAKE = list(K = PAKE, model = "RKHS")
)
# Modelo 37: E + W + GAK + PAK + GAKE + PAKE (Arc-cosseno)
Eta37 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
GAK = list(K = GAK, model = "RKHS"),
PAK = list(K = PAK, model = "RKHS"),
GAKE = list(K = GAKE, model = "RKHS"),
PAKE = list(K = PAKE, model = "RKHS")
)
# --- 2.3: Interação com Ambiente Climático (E + W + G/P + GxW/PxW) ---
# Modelo 38: E + W + G + GW (Linear)
Eta38 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
G = list(K = ZG, model = "RKHS"),
GW = list(K = ZGZW, model = "RKHS")
)
# Modelo 39: E + W + P + PW (Linear)
Eta39 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
P = list(K = ZP, model = "RKHS"),
PW = list(K = ZPZW, model = "RKHS")
)
# Modelo 40: E + W + G + P + GW + PW (Linear)
Eta40 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
G = list(K = ZG, model = "RKHS"),
P = list(K = ZP, model = "RKHS"),
GW = list(K = ZGZW, model = "RKHS"),
PW = list(K = ZPZW, model = "RKHS")
)
# Modelo 41: E + W + GGK + GGKW (Gaussiano)
Eta41 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
GGK = list(K = GGK, model = "RKHS"),
GGKW = list(K = GGKW, model = "RKHS")
)
# Modelo 42: E + W + PGK + PGKW (Gaussiano)
Eta42 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
PGK = list(K = PGK, model = "RKHS"),
PGKW = list(K = PGKW, model = "RKHS")
)
# Modelo 43: E + W + GGK + PGK + GGKW + PGKW (Gaussiano)
Eta43 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
GGK = list(K = GGK, model = "RKHS"),
PGK = list(K = PGK, model = "RKHS"),
GGKW = list(K = GGKW, model = "RKHS"),
PGKW = list(K = PGKW, model = "RKHS")
)
# Modelo 44: E + W + GAK + GAKW (Arc-cosseno)
Eta44 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
GAK = list(K = GAK, model = "RKHS"),
GAKW = list(K = GAKW, model = "RKHS")
)
# Modelo 45: E + W + PAK + PAKW (Arc-cosseno)
Eta45 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
PAK = list(K = PAK, model = "RKHS"),
PAKW = list(K = PAKW, model = "RKHS")
)
# Modelo 46: E + W + GAK + PAK + GAKW + PAKW (Arc-cosseno)
Eta46 <- list(
E = list(X = ZE, model = "BRR"),
W = list(K = ZW, "RKHS"),
GAK = list(K = GAK, model = "RKHS"),
PAK = list(K = PAK, model = "RKHS"),
GAKW = list(K = GAKW, model = "RKHS"),
PAKW = list(K = PAKW, model = "RKHS")
)
# -------------------------------------------------------------------
# GRUPO 3: MODELOS CLIMÁTICOS (EFEITO 'W' SUBSTITUI 'E')
# (Modelos Eta47 a Eta56)
# -------------------------------------------------------------------
# Modelo 47: W (Apenas kernel climático)
Eta47 <- list(
W = list(K = ZW, model = "RKHS")
)
# Modelo 48: W + G (Linear)
Eta48 <- list(
W = list(K = ZW, model = "RKHS"),
G = list(K = ZG, model = "RKHS")
)
# Modelo 49: W + P (Linear)
Eta49 <- list(
W = list(K = ZW, model = "RKHS"),
P = list(K = ZP, model = "RKHS")
)
# Modelo 50: W + G + P (Linear)
Eta50 <- list(
W = list(K = ZW, model = "RKHS"),
G = list(K = ZG, model = "RKHS"),
P = list(K = ZP, model = "RKHS")
)
# Modelo 51: W + GGK (Gaussiano)
Eta51 <- list(
W = list(K = ZW, model = "RKHS"),
GGK = list(K = GGK, model = "RKHS")
)
# Modelo 52: W + PGK (Gaussiano)
Eta52 <- list(
W = list(K = ZW, model = "RKHS"),
PGK = list(K = PGK, model = "RKHS")
)
# Modelo 53: W + GGK + PGK (Gaussiano)
Eta53 <- list(
W = list(K = ZW, model = "RKHS"),
GGK = list(K = GGK, model = "RKHS"),
PGK = list(K = PGK, model = "RKHS")
)
# Modelo 54: W + GAK (Arc-cosseno)
Eta54 <- list(
W = list(K = ZW, model = "RKHS"),
GAK = list(K = GAK, model = "RKHS")
)
# Modelo 55: W + PAK (Arc-cosseno)
Eta55 <- list(
W = list(K = ZW, model = "RKHS"),
PAK = list(K = PAK, model = "RKHS")
)
# Modelo 56: W + GAK + PAK (Arc-cosseno)
Eta56 <- list(
W = list(K = ZW, model = "RKHS"),
GAK = list(K = GAK, model = "RKHS"),
PAK = list(K = PAK, model = "RKHS")
)
# -------------------------------------------------------------------
# GRUPO 4: MODELOS DE EFEITOS INDIVIDUAIS (PARA COMPARAÇÃO)
# (Modelos Eta57 a Eta64)
# -------------------------------------------------------------------
# Modelo 57: E (Apenas efeito categórico de ambiente)
Eta57 <- list(
E = list(X = ZE, model = "BRR")
)
# Modelo 58: G (Apenas genômico linear)
Eta58 <- list(
G = list(K = ZG, model = "RKHS")
)
# Modelo 59: P (Apenas fenômico linear)
Eta59 <- list(
P = list(K = ZP, model = "RKHS")
)
# Modelo 60: GGK (Apenas genômico gaussiano)
Eta60 <- list(
GGK = list(K = GGK, model = "RKHS")
)
# Modelo 61: PGK (Apenas fenômico gaussiano)
Eta61 <- list(
PGK = list(K = PGK, model = "RKHS")
)
# Modelo 62: GAK (Apenas genômico arc-cosseno)
Eta62 <- list(
GAK = list(K = GAK, model = "RKHS")
)
# Modelo 63: PAK (Apenas fenômico arc-cosseno)
Eta63 <- list(
PAK = list(K = PAK, model = "RKHS")
)
# Modelo 64: W (Apenas kernel climático)
# Nota: Este é idêntico ao Eta47, incluído para consistência.
Eta64 <- list(
W = list(K = ZW, model = "RKHS")
)Para testar o Grupo 1 (Modelos Base E),
agrupamos os primeiros 18 modelos (Eta1 a
Eta18) em uma lista chamada Models. Esta lista
pode ser usada para uma análise preliminar focada apenas nos modelos GxE
categóricos.
# Cada modelo combina diferentes efeitos: genômico, fenômico, ambiente e interações
Models <- list(Eta1, Eta2, Eta3, Eta4, Eta5, Eta6, Eta7, Eta8, Eta9, Eta10,
Eta11, Eta12, Eta13, Eta14, Eta15, Eta16, Eta17, Eta18)
length(Models) # Deve retornar 18[1] 18Para rodar a validação cruzada com os novos modelos, adicione-os à
lista Models. Agora, vamos consolidar
todos os modelos para a validação cruzada completa.
# 1. Primeiro, agrupamos todos os modelos do Grupo 2 e Grupo 3
# (aqueles que usam o kernel climático W) na lista `Climate_Models`.
Climate_Models <- list(Eta20, Eta21, Eta22, Eta23, Eta24, Eta25, Eta26, Eta27, Eta28,
Eta29, Eta30, Eta31, Eta32, Eta33, Eta34, Eta35, Eta36, Eta37,
Eta38, Eta39, Eta40, Eta41, Eta42, Eta43, Eta44, Eta45, Eta46,
Eta48, Eta49, Eta50, Eta51, Eta52, Eta53, Eta54, Eta55, Eta56)
# 2. Adicionamos os modelos de efeitos individuais (Grupo 4)
Climate_Models <- c(Climate_Models, list(Eta57, Eta58, Eta59, Eta60, Eta61, Eta62, Eta63, Eta64))
# 3. Finalmente, combinamos a lista original `Models` (Grupo 1) com a `Climate_Models`
# para criar o "menu" completo de modelos.
Models_full_set <- c(Models, Climate_Models)
# Esta lista `Models_full_set` está pronta para ser usada no loop
# de validação cruzada, permitindo uma comparação completa de todos os 62 modelos.
cat("Total de modelos a serem avaliados:", length(Models_full_set))Total de modelos a serem avaliados: 6213) Cross-validação
Esta é a seção de testes do projeto. O objetivo é avaliar rigorosamente a capacidade preditiva de todos os nossos modelos (Eta1 a Eta64) para determinar quais deles são realmente úteis.
Um modelo que apenas se ajusta bem aos dados existentes (bom fit) não é útil; ele precisa ser capaz de prever dados que nunca viu. Para simular isso, usamos a Validação Cruzada (CV).
Vamos implementar múltiplos esquemas de CV para simular diferentes desafios do mundo real no melhoramento:
- CV1: validação por genótipo (testa genótipos não
observados).
- Pergunta: O modelo pode prever o desempenho de um genótipo (híbrido) completamente novo, que não estava em nenhum ambiente de treinamento?
- CV2: validação por célula (testa combinações genótipo ×
ambiente parcialmente observadas).
- Pergunta: O modelo pode prever o desempenho de um genótipo conhecido em um ambiente novo para ele (assumindo que o genótipo foi visto em outros ambientes e o ambiente foi visto com outros genótipos)?
- CV0: leave-one-environment-out (testa ambientes inteiros não
observados).
- Pergunta: O teste mais difícil. O modelo pode prever o desempenho de todos os genótipos em um ambiente completamente novo (um ano ou local que o modelo nunca viu)?
- CV00: leave-one-environment-out (testa genótipos em
ambientes não observados).
- Semelhante ao CV0, focado em prever genótipos dentro de um ambiente não visto.
13.0) Visualização dos esquemas de validação cruzada
# ------------------------------------------------------------
# Parâmetros gerais
# ------------------------------------------------------------
n_geno <- 50 # número de genótipos
n_env <- 4
envs <- paste0("Env ", 1:n_env)
# ------------------------------------------------------------
# Função para gerar estrutura de cada esquema CV
# ------------------------------------------------------------
generate_cv_data <- function(type) {
df <- expand.grid(Genotype = 1:n_geno, Env = envs)
df$Test <- FALSE
if (type == "CV2") {
# 20% dos genótipos testados (genótipo parcialmente testado)
test_genos <- sample(1:nrow(df), nrow(df) * 0.2)
df$Test[test_genos] <- TRUE
} else if (type == "CV1") {
# 20% dos genótipos totalmente não vistos
test_genos <- sample(1:n_geno, n_geno * 0.2)
df$Test[df$Genotype %in% test_genos] <- TRUE
} else if (type == "CV0") {
# Um ambiente completamente novo (Env 2)
df$Test[df$Env == "Env 2"] <- TRUE
} else if (type == "CV00") {
# Genótipos e ambiente totalmente novos (sem esses genótipos em outros ambientes)
test_genos <- sample(1:n_geno, n_geno * 0.2)
df$Test <- FALSE
df <- df %>%
mutate(Test = ifelse(Env == "Env 2" & Genotype %in% test_genos, TRUE, FALSE)) %>%
filter(!(Genotype %in% test_genos & Env != "Env 2")) %>% # remove genótipos de teste dos outros ambientes
filter(!(Env == "Env 2" & !(Genotype %in% test_genos))) # mantém apenas genótipos de teste em Env 2
}
# garantir ordem correta dos ambientes
df$CV <- type
df
}
# ------------------------------------------------------------
# Gerar dados
# ------------------------------------------------------------
cv_types <- c("CV2", "CV0", "CV1", "CV00") # ordem original
data <- do.call(rbind, lapply(cv_types, generate_cv_data))
# ------------------------------------------------------------
# Função para criar gráfico individual
# ------------------------------------------------------------
plot_cv <- function(df, title) {
ggplot(df, aes(x = as.character(Env), y = Genotype)) +
# Linhas azuis finas de fundo apenas para pontos de treino
geom_tile(data = subset(df, !Test), fill = "#5dade2", height = 0.4, width = 0.9) +
# Pontos pretos para observações de teste
geom_point(data = subset(df, Test), color = "black", size = 2) +
theme_minimal(base_size = 12) +
theme(
axis.title.x = element_blank(),
axis.title.y = element_blank(),
axis.text.x = element_text(size = 10),
axis.text.y = element_blank(),
panel.grid = element_blank(),
plot.title = element_text(hjust = 0.5, face = "bold", size = 14),
legend.position = "none"
) +
labs(title = title)
}
# ------------------------------------------------------------
# Criar gráficos
# ------------------------------------------------------------
p1 <- plot_cv(filter(data, CV == "CV2"), "CV2")
p2 <- plot_cv(filter(data, CV == "CV0"), "CV0")
p3 <- plot_cv(filter(data, CV == "CV1"), "CV1")
p4 <- plot_cv(filter(data, CV == "CV00"), "CV00")
# ------------------------------------------------------------
# Combinar gráficos com eixos globais
# ------------------------------------------------------------
grid_arrange_shared_legend <- function(..., ncol = 2, nrow = 2,
x_label = "Environments",
y_label = "Genotypes") {
plots <- list(...)
grid <- arrangeGrob(grobs = plots, ncol = ncol, nrow = nrow)
grid <- arrangeGrob(
grid,
bottom = textGrob(x_label, gp = gpar(fontsize = 14)),
left = textGrob(y_label, rot = 90, gp = gpar(fontsize = 14))
)
grid
}
final_plot <- grid_arrange_shared_legend(p1, p2, p3, p4)
grid.newpage()
grid.draw(final_plot)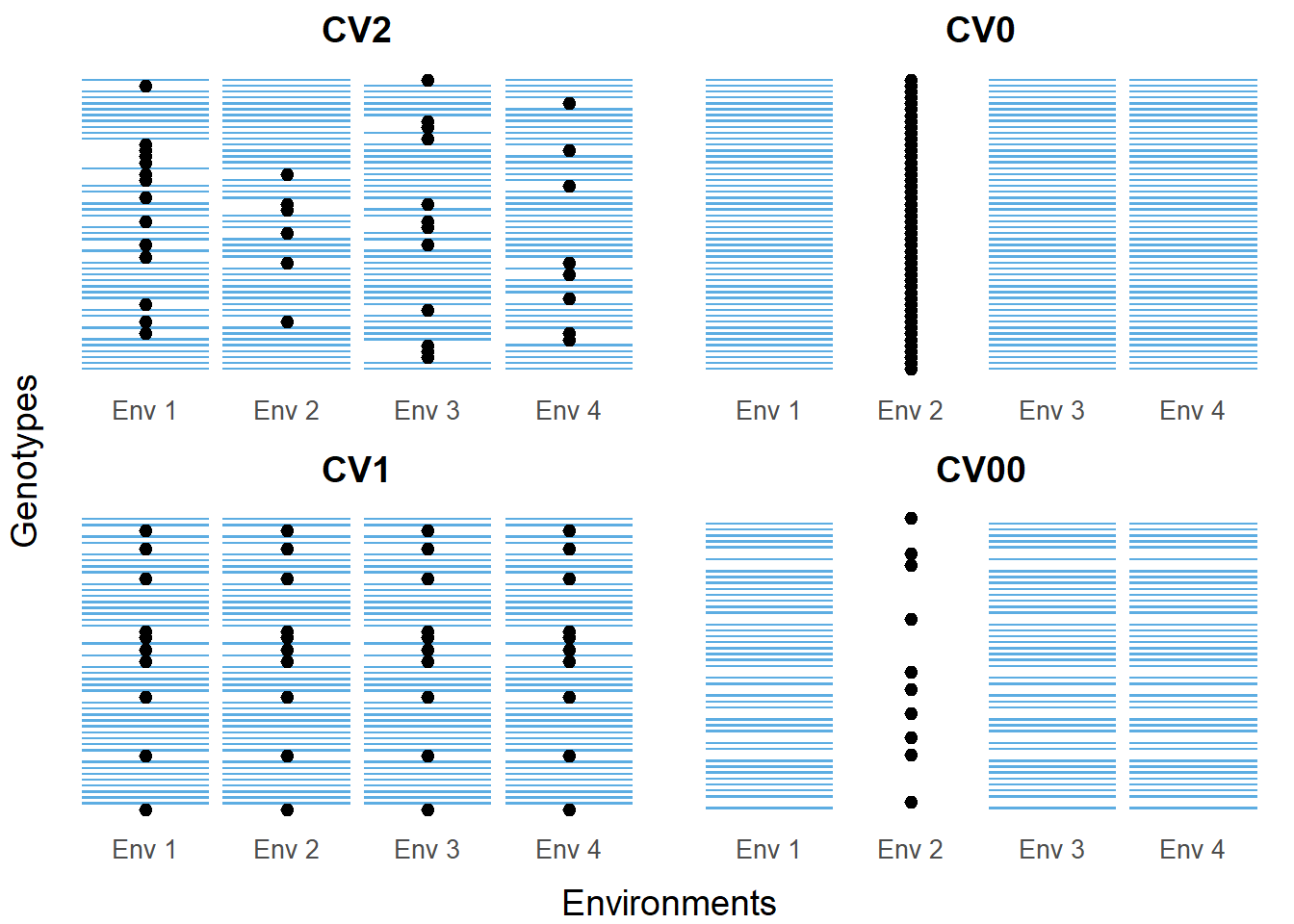
| Version | Author | Date |
|---|---|---|
| df8457c | WevertonGomesCosta | 2025-11-04 |
ggsave("output/cv-schemes.tiff", final_plot, width = 10, height = 8)13.1) Parâmetros para execução dos modelos
Aqui, definimos os parâmetros tanto para o motor do BGLR
(o amostrador MCMC) quanto para a estrutura da nossa validação
cruzada.
# Parâmetros de execução do BGLR
nIter <- 5000 # número total de iterações da cadeia MCMC
burnIn <- 1000 # número de iterações descartadas como burn-in
thin <- 10 # intervalo de amostragem para reduzir autocorrelação
num_rep <- 20 # número de repetições da validação cruzada
num_fold <- 5 # número de folds em cada repetiçãonIter = 5000: O número total de “passos” que o algoritmo Bayesiano (MCMC) dará para estimar os parâmetros.burnIn = 1000: O número de passos iniciais que descartamos. Isso dá tempo ao modelo para “aquecer” e convergir para uma solução estável.thin = 10: Para economizar memória, só guardamos 1 em cada 10 passos (após o burn-in). Teremos (5000-1000)/10 = 400 amostras por parâmetro.num_rep = 20: Repetimos todo o processo de CV 20 vezes (com diferentes embaralhamentos aleatórios) para garantir que nossos resultados de acurácia não sejam devidos ao acaso.num_fold = 5: Em cada repetição, dividimos os dados em 5 partes (“folds”). O modelo treina em 4 partes (80% dos dados) e é testado na 1 parte restante (20%).
13.2) Verificações rápidas
Esta é uma verificação de sanidade (sanity check). A
execução da validação cruzada pode levar horas ou dias. Este bloco atua
como uma trava de segurança
(stopifnot).
Ele verifica se os objetos de dados fundamentais (Pheno,
Pedigree) e as colunas de traits (GY,
KW) estão presentes na memória. Se algo estiver faltando, o
script irá parar agora com um erro claro, em vez de falhar horas depois
no meio do processamento paralelo.
# Verificações básicas de consistência dos dados
if (!exists("Pheno")) stop("Objeto `Pheno` não encontrado no ambiente.")
if (!"Pedigree" %in% names(Pheno)) stop("Coluna 'Pedigree' não encontrada em Pheno.")
if (!"GY" %in% names(Pheno)) stop("Coluna 'GY' não encontrada em Pheno (resposta).")
if (!"KW" %in% names(Pheno)) stop("Coluna 'KW' não encontrada em Pheno (resposta).")
# Se 'Pedigree' não existir como objeto, cria a partir de Pheno
if (!exists("Pedigree")) Pedigree <- unique(Pheno$Pedigree)
# Garante que não haja duplicatas
Pedigree <- unique(Pedigree)
# Número total de híbridos consistentes
n_hybrids <- length(Pedigree)
# Verificação extra: não pode ser zero
stopifnot(n_hybrids > 0)13.3) Pastas de saída
Vamos gerar milhares de arquivos de resultado (um para cada
fold de cada rep de cada modelo). Este bloco
de código simplesmente garante que as pastas de destino
(output/results e
output/componentes_variancia) existam.
recursive = TRUE cria a estrutura de pastas necessária
(ex: output/ e output/results/ de uma só vez),
e showWarnings = FALSE evita mensagens desnecessárias caso
as pastas já tenham sido criadas.
# Cria diretórios de saída para armazenar resultados
dir.create("output/results", recursive = TRUE, showWarnings = FALSE)13.5) Funções auxiliares e ambientes de CV0/CV00
Este bloco prepara as “ferramentas” que usaremos dentro do nosso loop de processamento paralelo.
envs_leaveout: Define os ambientes específicos que serão usados para os cenários de teste CV0 e CV00 (onde deixamos um ambiente inteiro de fora).run_bglr: Esta é uma função ‘wrapper’ (empacotadora) personalizada e altamente otimizada. A funçãoBGLRpadrão retorna objetos muito pesados. Para evitar sobrecarregar a memória em um loop paralelo, nossarun_bglrfaz o seguinte:- Roda o
BGLR. - Salva os arquivos de log do MCMC (via
saveAt). - Extrai apenas as métricas essenciais (predições
yHat, variâncias médias) e as salva em um único arquivo.rdsleve (_metricas.rds). - Limpa o objeto
fitpesado da memória (rm(fit); gc()). - Retorna apenas o
yHat(predições) e o tempo de execução para o loop principal.
- Roda o
calc_cor_by_env: Uma função auxiliar simples que usaremos após a execução principal, para agregar os resultados e calcular a acurácia (correlação) dentro de cada ambiente.combos: Esta é a “lista de tarefas” mestra para o processamento paralelo.expand.gridcria uma tabela gigante onde cada linha é um “trabalho” (uma combinação única deMODELO,REPETIÇÃOeTRAIT). O loopforeach(na próxima seção) irá distribuir essas linhas pelos núcleos da CPU.
# Ambientes a serem deixados de fora (leave-one-environment-out)
envs_leaveout <- c("CS11_WS", "CS11_WW", "CS12_WS", "CS12_WW")
# --- FUNÇÃO OTIMIZADA ---
# Função auxiliar: roda BGLR, salva MÉTRICAS RESUMIDAS e retorna predições + tempo
run_bglr <- function(y2_vector, ETA, nIter, burnIn, thin, save_prefix) {
# y2_vector: vetor de resposta com NAs nos pontos a predizer
y_t <- as.numeric(y2_vector)
dir.create(dirname(save_prefix), recursive = TRUE, showWarnings = FALSE)
# mede tempo
start_time <- Sys.time()
fit <- BGLR(y = y_t, ETA = ETA, nIter = nIter, burnIn = burnIn, thin = thin, saveAt = save_prefix)
end_time <- Sys.time()
elapsed <- as.numeric(difftime(end_time, start_time, units = "secs"))
# 1. Extrair componentes de variância (varU) de forma dinâmica
# Isto funciona para QUALQUER modelo (com 1 ou N efeitos)
variancias_U <- list()
if (!is.null(fit$ETA)) {
nomes_efeitos <- names(fit$ETA)
if (is.null(nomes_efeitos)) {
nomes_efeitos <- paste0("Efeito_", seq_along(fit$ETA))
}
for (i in seq_along(fit$ETA)) {
efeito_nome <- nomes_efeitos[i]
# Armazena a variância média (pós-burnIn)
if (!is.null(fit$ETA[[i]]$varU)) {
variancias_U[[efeito_nome]] <- fit$ETA[[i]]$varU
}
}
}
# 2. Criar o objeto de saída leve
saida_essencial <- list(
yHat_mean = fit$yHat, # Predições (média)
varE_mean = fit$varE, # Variância residual (média)
# --- LINHA ADICIONADA ---
# Salva a variância dos efeitos fixos (varB), se ela existir
varB_mean = ifelse(is.null(fit$varB), NA, fit$varB),
# --- FIM DA LINHA ADICIONADA ---
variances_U_mean = variancias_U, # Lista de variâncias dos kernels (média)
DIC = ifelse(is.null(fit$fit$DIC), NA, fit$fit$DIC),
runtime_sec = elapsed
)
# 3. Salvar este objeto de métricas leve (arquivo .rds minúsculo)
# Isso substitui o saveRDS(fit, ...)
saveRDS(saida_essencial, paste0(save_prefix, "_metricas.rds"))
# --- FIM DAS MUDANÇAS ---
# 4. Preparar o retorno para o 'foreach' (isto permanece igual)
# O loop 'foreach' só precisa do yHat e do runtime
retorno_foreach <- list(yHat = fit$yHat, runtime_sec = elapsed)
# 5. Limpar o objeto 'fit' pesado da memória imediatamente
rm(fit, saida_essencial, variancias_U)
gc() # Força a "coleta de lixo"
# retorna lista com predições e tempo
return(retorno_foreach)
}
# --- FIM DA FUNÇÃO OTIMIZADA ---
# Função auxiliar: calcula correlação observada vs predita por ambiente
# (Esta função não precisa de alteração)
calc_cor_by_env <- function(df_with_yhat, trait_col) {
df_with_yhat %>%
dplyr::group_by(Env) %>%
dplyr::summarize(
cor = cor(.data[[trait_col]], yhat, use = "complete.obs"),
.groups = "drop"
) %>%
as.data.frame()
}
# Cria grid de combinações: cada worker processa 1 modelo × 1 repetição
# (Esta função não precisa de alteração)
combos <- expand.grid(MODEL = 1:length(Models_full_set), REP = seq_len(num_rep), TRAIT = c("GY", "KW"))Esta é a limpeza final antes do loop de
processamento principal. Usamos setdiff(ls(), ...) para
obter uma lista de tudo o que está no ambiente, exceto
os objetos essenciais que listamos (Pheno,
Models_full_set, nossas funções, etc.).
Em seguida, rm() remove todo esse “lixo” intermediário.
Isso garante que cada “trabalhador” paralelo (núcleo de CPU) seja
iniciado com o mínimo de memória possível, dedicando todos os recursos
ao ajuste do BGLR.
# Excluindo todos os objetos intermediários para liberar memória
rm(list = setdiff(ls(), c("Pheno", "Pedigree", "Models_full_set",
"nIter", "burnIn", "thin",
"num_rep", "num_fold",
"envs_leaveout",
"run_bglr", "calc_cor_by_env",
"combos", "n_hybrids", "model_info")))14) Ajuste de modelos
Esta seção é o “motor” de toda a análise. Aqui, executamos o plano de validação cruzada definido na Seção 13.
14.1) Configuração do backend paralelo
Por que isso é necessário? Temos uma tarefa computacional enorme pela frente. O número total de modelos a ajustar é:
64 Modelos × 2 Traits ×
20 Repetições × 5 Folds = 12.800
execuções do BGLR (para CV1/CV2)
…mais um número ainda maior para CV0/CV00. Fazer isso em um único núcleo de CPU levaria semanas.
Este bloco de código detecta quantos núcleos de processamento
(cores) seu computador possui e reserva (quase) todos eles
(useCores) para o trabalho.
doParallel::registerDoParallel(cl) informa ao R para usar
este “cluster” de núcleos para executar tarefas simultaneamente,
acelerando drasticamente o processo.
# Detecta número de núcleos disponíveis e configura backend paralelo
numCores <- parallel::detectCores()
useCores <- max(1, numCores - 1) # usa todos menos 1 núcleo
cl <- parallel::makeCluster(useCores)
doParallel::registerDoParallel(cl)
cat("Rodando em ", useCores, " núcleos.")Rodando em 21 núcleos.gc() é chamado para “coletar o lixo” (liberar memória
RAM não utilizada) antes de iniciar o trabalho pesado.
gc() # Limpa memória antes da execução paralela used (Mb) gc trigger (Mb) max used (Mb)
Ncells 1844467 98.6 3602795 192.5 2516773 134.5
Vcells 21513541 164.2 140582013 1072.6 219659394 1675.914.2) Execução dos modelos em paralelo
Este é o coração da análise. O loop foreach
itera sobre cada linha da nossa “lista de tarefas” mestra
(combos). Graças ao doParallel, cada iteração
(um “trabalho”) é enviada para um núcleo de CPU diferente para ser
executada simultaneamente.
Dentro de cada iteração paralela (para cada núcleo):
- Identificar o Trabalho: O worker
identifica qual
MODEL,rep_numeTRAITele deve processar (ex: Modelo 10, Repetição 5, Trait “GY”). - Criar Folds (CV1/CV2): O worker cria os 5
folds (partições) para os genótipos.
train_genoetest_genosão definidos. - Loop Interno (por Fold): O worker então
itera através de cada um dos 5 folds.
- Mascarar Dados (CV1/CV2): Ele cria
yield$Y2. Este vetor é uma cópia dos fenótipos onde todas as observações dos genótipos emtest_genosão definidas comoNA. - Ajustar Modelo (CV1/CV2): O
BGLRé chamado usandoyield$Y2como resposta. OBGLRirá “preencher” (prever) os valoresNA. - Coletar Resultados (CV1/CV2): As predições
(
yhat) são extraídas.df_testcontém as predições para os genótipos de validação (que eramNA).df_traincontém as predições para os genótipos de treinamento. A acurácia (correlação) é calculada para ambos e armazenada emCV1_listeCV2_list. - Mascarar Dados (CV0/CV00): Um segundo conjunto de
máscaras é criado (
yield2$Y2). Ele é semelhante ao primeiro, mas também define comoNAtodas as observações de um ambiente específico (ex: “CS11_WS”). - Ajustar Modelo (CV0/CV00): O
BGLRé executado novamente com esta nova máscara. - Coletar Resultados (CV0/CV00): A acurácia é
calculada e armazenada em
CV0_listseCV00_lists.
- Mascarar Dados (CV1/CV2): Ele cria
- Salvar Resultados: Após o loop de 5
folds terminar, o worker agrega os resultados de
acurácia (ex:
CV1out) e os salva em um arquivo.csvúnico para aqueleMODEL,REPeTRAIT(ex:CV1_GY_Eta10_rep5.csv). - O worker então se torna livre para pegar o próximo
“trabalho” da fila
combos.
Nota: Este bloco está com
eval=FALSE para que o tutorial possa ser compilado
rapidamente. Para executar a análise completa, mude a opção do chunk
para {r parallel-model, cache=FALSE, eval=TRUE} e
execute-o. Isso levará um tempo considerável.
# Paraleliza
foreach(i = seq_len(nrow(combos)),
.packages = c("BGLR", "dplyr", "plyr")) %dopar% {
MODEL <- combos$MODEL[i]
rep_num <- combos$REP[i]
TRAIT <- as.character(combos$TRAIT[i])
pheno <- data.frame(Pheno) # copia de Pheno para manipulação local
# Diretórios organizados por repetição e modelo
rep_dir <- file.path("output/ajuste_modelo", paste0("rep_", rep_num))
dir.create(rep_dir, recursive = TRUE, showWarnings = FALSE)
eta_dir <- file.path(rep_dir, paste0("Eta", MODEL))
dir.create(eta_dir, recursive = TRUE, showWarnings = FALSE)
# Inicializa listas de resultados
CV1_list <- list()
CV2_list <- list()
CV0_lists <- lapply(envs_leaveout, function(x)
list())
CV00_lists <- lapply(envs_leaveout, function(x)
list())
names(CV0_lists) <- envs_leaveout
names(CV00_lists) <- envs_leaveout
# Cria folds reprodutíveis
#set.seed(1000 + MODEL * 100 + rep_num)
obs_per_fold <- ceiling(n_hybrids / num_fold)
folds <- sample(rep(seq_len(num_fold), each = obs_per_fold, length.out = n_hybrids))
folds_df <- data.frame(Pedigree = Pedigree,
fold = folds,
stringsAsFactors = FALSE)
for (fold_num in seq_len(num_fold)) {
train_geno <- folds_df$Pedigree[folds_df$fold != fold_num]
test_geno <- folds_df$Pedigree[folds_df$fold == fold_num]
# --- CV1 / CV2 ---
yield <- pheno
yield$Y2 <- NA
yield$Y2[yield$Pedigree %in% train_geno] <- yield[[TRAIT]][yield$Pedigree %in% train_geno]
save_prefix_full <- file.path(eta_dir,
paste0("full_Eta", MODEL, "_", TRAIT, "_fold", fold_num, "_"))
#set.seed(2000 + MODEL * 100 + rep_num * 10 + fold_num)
res_full <- run_bglr(
yield$Y2,
ETA = Models_full_set[[MODEL]],
nIter = nIter,
burnIn = burnIn,
thin = thin,
save_prefix_full
)
yield$yhat <- res_full$yHat
runtime_full <- res_full$runtime_sec
df_test <- yield[!yield$Pedigree %in% train_geno, ]
df_train <- yield[yield$Pedigree %in% train_geno, ]
tmp1 <- calc_cor_by_env(df_test, TRAIT)
tmp2 <- calc_cor_by_env(df_train, TRAIT)
tmp1$Runtime_sec <- runtime_full
tmp2$Runtime_sec <- runtime_full
CV1_list[[length(CV1_list) + 1]] <- tmp1
CV2_list[[length(CV2_list) + 1]] <- tmp2
# --- CV0 / CV00 ---
for (env_idx in seq_along(envs_leaveout)) {
env_name <- envs_leaveout[env_idx]
yield2 <- pheno
yield2$Y2 <- NA
yield2$Y2[yield2$Pedigree %in% train_geno] <- yield2[[TRAIT]][yield2$Pedigree %in% train_geno]
yield2$Y2[yield2$Env == env_name] <- NA
save_prefix_env <- file.path(eta_dir,
paste0(env_name, "_", TRAIT, "_Eta", MODEL, "_fold", fold_num, "_"))
#set.seed(3000 + MODEL * 100 + rep_num * 10 + fold_num + env_idx)
res_env <- run_bglr(
yield2$Y2,
ETA = Models_full_set[[MODEL]],
nIter = nIter,
burnIn = burnIn,
thin = thin,
save_prefix_env
)
yield2$yhat <- res_env$yHat
runtime_env <- res_env$runtime_sec
df_test2 <- yield2[!yield2$Pedigree %in% train_geno, ]
df_train2 <- yield2[yield2$Pedigree %in% train_geno, ]
tmp00 <- calc_cor_by_env(df_test2, TRAIT)
tmp0 <- calc_cor_by_env(df_train2, TRAIT)
tmp00$Runtime_sec <- runtime_env
tmp0$Runtime_sec <- runtime_env
CV00_lists[[env_idx]][[length(CV00_lists[[env_idx]]) + 1]] <- tmp00
CV0_lists[[env_idx]][[length(CV0_lists[[env_idx]]) + 1]] <- tmp0
}
}
# Salva resultados por esquema
CV1out <- plyr::ldply(CV1_list, data.frame)
CV2out <- plyr::ldply(CV2_list, data.frame)
write.csv(CV1out,
file = file.path(
"output/results",
paste0("CV1_", TRAIT, "_Eta", MODEL, "_rep", rep_num, ".csv")
),
row.names = FALSE)
write.csv(CV2out,
file = file.path(
"output/results",
paste0("CV2_", TRAIT, "_Eta", MODEL, "_rep", rep_num, ".csv")
),
row.names = FALSE)
for (env_idx in seq_along(envs_leaveout)) {
env_name <- envs_leaveout[env_idx]
CV00out <- plyr::ldply(CV00_lists[[env_idx]], data.frame)
CV0out <- plyr::ldply(CV0_lists[[env_idx]], data.frame)
write.csv(CV00out,
file = file.path(
"output/results",
paste0(
"CV00_",
TRAIT,
"_",
env_name,
"_Eta",
MODEL,
"_rep",
rep_num,
".csv"
)
),
row.names = FALSE)
write.csv(CV0out,
file = file.path(
"output/results",
paste0(
"CV0_",
TRAIT,
"_",
env_name,
"_Eta",
MODEL,
"_rep",
rep_num,
".csv"
)
),
row.names = FALSE)
}
NULL
} # end foreachApós o término do loop foreach, este comando
desliga o cluster paralelo e libera os núcleos da CPU
de volta para o sistema operacional.
15) Resultados
O loop de processamento paralelo na Seção 14 gerou
milhares de arquivos .csv individuais (um
para cada Modelo x Repetição x
Trait x Fold x CV). Esta seção é
o pós-processamento, onde iremos:
- Consolidar: Ler todos esses arquivos pequenos e agrupá-los em alguns arquivos “mestres”.
- Limpar: Os arquivos “mestres” ainda estarão “sujos”, pois toda a informação importante (Qual modelo? Qual trait?) está “presa” no nome do arquivo.
- Analisar: Vamos “limpar” esses arquivos mestres,
extraindo (fazendo parsing) os nomes dos arquivos em colunas de
metadados (ex:
Model,Trait,Rep).
O resultado final serão alguns arquivos .csv limpos e
prontos para a visualização e análise estatística.
15.1) Segregação dos resultados dos esquemas CV00 e CV0 por ambiente
Objetivo: Consolidar os resultados dos cenários leave-one-environment-out.
Como o “ambiente-deixado-de-fora” é o fator chave aqui, primeiro agrupamos os resultados por esse fator.
- O código lista todos os arquivos
CV0_eCV00_. - Em seguida, ele filtra essa lista para um ambiente-alvo específico
(ex:
grep("CS12_WS", ...)). - Ele lê todos os arquivos
.csvdesse subconjunto (ex: todos os resultados de CV0 onde “CS12_WS” foi o ambiente de teste) e os combina em um único dataframe (ex:df_CV0_CS12_WS). - Esse dataframe consolidado é salvo como um “resumo
intermediário” (ex:
Pred.ability.CV0.CS12_WS.csv). - Isso é repetido para todos os 4 ambientes e 2 tipos de CV (8 resumos intermediários).
- Finalmente, todos esses resumos intermediários são combinados em um
único arquivo mestre (
df_CV0_CV00).
# Selecionando apenas os que contêm "CV0"
list_csv_files_CV0 <- list.files(
path = "output/results",
pattern = "CV0_",
# pega arquivos que contenham CV0
full.names = TRUE
)
# Selecionando apenas os que contêm "CV00"
list_csv_files_CV00 <- list.files(
path = "output/results",
pattern = "CV00_",
# pega arquivos que contenham CV0
full.names = TRUE
)
# Filtrando apenas os arquivos que contêm "CS12_WS"
list_csv_files_CV0_CS12_WS <- list_csv_files_CV0[grep("CS12_WS", list_csv_files_CV0)]
df_CV0_CS12_WS <- read_csv(list_csv_files_CV0_CS12_WS, id = "file_name") %>% as.data.frame()
head(df_CV0_CS12_WS)
write.csv(df_CV0_CS12_WS, "output/Pred.ability/Pred.ability.CV0.CS12_WS.csv") # Salva consolidação
list_csv_files_CV00_CS12_WS <- list_csv_files_CV00[grep("CS12_WS", list_csv_files_CV00)]
df_CV00_CS12_WS <- read_csv(list_csv_files_CV00_CS12_WS, id = "file_name") %>% as.data.frame()
head(df_CV00_CS12_WS)
write.csv(df_CV00_CS12_WS, "output/Pred.ability/Pred.ability.CV00.CS12_WS.csv") # Salva consolidação
# Filtrando apenas os arquivos que contêm "CS11_WW"
list_csv_files_CV0_CS11_WW <- list_csv_files_CV0[grep("CS11_WW", list_csv_files_CV0)]
df_CV0_CS11_WW <- read_csv(list_csv_files_CV0_CS11_WW, id = "file_name") %>% as.data.frame()
head(df_CV0_CS11_WW)
write.csv(df_CV0_CS11_WW, "output/Pred.ability/Pred.ability.CV0.CS11_WW.csv") # Salva consolidação
list_csv_files_CV00_CS11_WW <- list_csv_files_CV00[grep("CS11_WW", list_csv_files_CV00)]
df_CV00_CS11_WW <- read_csv(list_csv_files_CV00_CS11_WW, id = "file_name") %>% as.data.frame()
head(df_CV00_CS11_WW)
write.csv(df_CV00_CS11_WW, "output/Pred.ability/Pred.ability.CV00.CS11_WW.csv") # Salva consolidação
# Filtrando apenas os arquivos que contêm "CS12_WW"
list_csv_files_CV0_CS12_WW <- list_csv_files_CV0[grep("CS12_WW", list_csv_files_CV0)]
df_CV0_CS12_WW <- read_csv(list_csv_files_CV0_CS12_WW, id = "file_name") %>% as.data.frame()
head(df_CV0_CS12_WW)
write.csv(df_CV0_CS12_WW, "output/Pred.ability/Pred.ability.CV0.CS12_WW.csv") # Salva consolidação
list_csv_files_CV00_CS12_WW <- list_csv_files_CV00[grep("CS12_WW", list_csv_files_CV00)]
df_CV00_CS12_WW <- read_csv(list_csv_files_CV00_CS12_WW, id = "file_name") %>% as.data.frame()
head(df_CV00_CS12_WW)
write.csv(df_CV00_CS12_WW, "output/Pred.ability/Pred.ability.CV00.CS12_WW.csv") # Salva consolidação
# Filtrando apenas os arquivos que contêm "CS11_WS"
list_csv_files_CV0_CS11_WS <- list_csv_files_CV0[grep("CS11_WS", list_csv_files_CV0)]
df_CV0_CS11_WS <- read_csv(list_csv_files_CV0_CS11_WS, id = "file_name") %>% as.data.frame()
head(df_CV0_CS11_WS)
write.csv(df_CV0_CS11_WS, "output/Pred.ability/Pred.ability.CV0.CS11_WS.csv") # Salva consolidação
list_csv_files_CV00_CS11_WS <- list_csv_files_CV00[grep("CS11_WS", list_csv_files_CV00)]
df_CV00_CS11_WS <- read_csv(list_csv_files_CV00_CS11_WS, id = "file_name") %>% as.data.frame()
head(df_CV00_CS11_WS)
write.csv(df_CV00_CS11_WS, "output/Pred.ability/Pred.ability.CV00.CS11_WS.csv") # Salva consolidação
df_CV0_CV00 <- bind_rows(
df_CV0_CS12_WS,
df_CV00_CS12_WS,
df_CV0_CS11_WW,
df_CV00_CS11_WW,
df_CV0_CS12_WW,
df_CV00_CS12_WW,
df_CV0_CS11_WS,
df_CV00_CS11_WS
)
write.csv(df_CV0_CV00, "output/Pred.ability/Pred.ability.CV0.CV00.csv") # Salva consolidação15.2) Resultados CV0 e CV00
Objetivo: Limpar e padronizar o arquivo mestre
CV0.CV00.csv.
Este bloco lê o arquivo mestre (...CV0.CV00.csv) criado
na etapa anterior e o limpa. O arquivo original é
“sujo” porque toda a informação útil (qual modelo, qual trait) está
“presa” dentro da coluna file_name.
- Usa
separatepara quebrar o nome do arquivo (ex:CV0_GY_CS11_WS_Eta1_rep1) em colunas limpas:CV,Trait,Env_Val(combinandoEnv1eEnv2),Model,Rep. sub("^Eta", "M", ...)padroniza o nome do modelo (ex: ‘Eta1’ -> ‘M1’) para facilitar a criação de gráficos.- Seleciona apenas as colunas úteis e salva o dataframe limpo, substituindo o arquivo antigo. O arquivo agora está pronto para análise.
# Agrupa por modelo, tipo de validação e traço
# Calcula média e desvio padrão da correlação preditiva
df <- read.csv("output/Pred.ability/Pred.ability.CV0.CV00.csv") %>%
mutate(
file_name = basename(file_name),
# remove caminho
file_name = sub("\\.csv$", "", file_name)
) %>% # remove extensão
# Quebra em até 6 partes (CV, Trait, Env1, Env2, ModelRaw, Rep)
separate(
file_name,
into = c("CV", "Trait", "Env1", "Env2", "ModelRaw", "Rep"),
sep = "_",
fill = "right",
# preenche com NA se faltar
remove = FALSE
) %>%
# Cria coluna Env apenas se houver Env1 e Env2
mutate(
Env_Val = paste0(Env1, "_", Env2),
Model = sub("^Eta", "M", sub("\\.csv$", "", ModelRaw))) %>%
dplyr::select(Model, CV, Trait, Env_Val, Env, Rep, cor) # remove colunas auxiliares
write.csv(df, "output/Pred.ability/Pred.ability.CV0.CV00.classified.csv") # Salva consolidação15.3) Consolidação dos resultados dos esquemas CV1 e CV2
Objetivo: Consolidar os resultados dos cenários CV1 (predição de genótipos novos) e CV2 (predição de genótipos em ambientes novos).
Este processo é mais simples que o de CV0/CV00 porque não precisamos agrupar por ambiente-deixado-de-fora.
list.filesencontra todos os arquivosCV1ouCV2.read_csvlê todos os milhares de arquivos de uma só vez (usando o argumentoid = "file_name"para rastrear a origem) e os combina em um único dataframe mestre,df_CV1_CV2.- Salva este dataframe mestre.
# Lê os arquivos de predição dos esquemas CV1 e CV2
list_csv_files <- list.files(
path = "output/results",
pattern = "CV1|CV2",
# pega arquivos que contenham CV1 ou CV2
full.names = TRUE
)
df_CV1_CV2 <- read_csv(list_csv_files, id = "file_name") %>% as.data.frame()
head(df_CV1_CV2)
write.csv(df_CV1_CV2, "output/Pred.ability/Pred.ability.CV1.CV2.csv") # Salva consolidação15.4) Resultados CV1 e CV2
Objetivo: Limpar e padronizar o arquivo mestre
CV1.CV2.csv.
Assim como na seção 15.2, este bloco limpa o arquivo
mestre ...CV1.CV2.csv que acabamos de criar. O arquivo
original é “sujo” porque toda a informação útil (qual modelo, qual
trait) está “presa” dentro da coluna file_name.
- Usa
separatepara quebrar o nome do arquivo (ex:CV1_GY_Eta1_rep1) em colunas limpas:CV,Trait,Model,Rep. (Note que não háEnvno nome do arquivo aqui, pois CV1/CV2 são calculados dentro dos ambientes). - Padroniza os nomes dos modelos (ex: ‘Eta1’ -> ‘M1’) para facilitar a criação de gráficos.
- Salva o dataframe limpo e “pronto para análise”, substituindo o arquivo antigo.
# Agrupa por modelo, tipo de validação e traço
# Calcula média e desvio padrão da correlação preditiva
df <- read.csv("output/Pred.ability/Pred.ability.CV1.CV2.csv") %>%
mutate(
file_name = basename(file_name),
# remove caminho
file_name = sub("\\.csv$", "", file_name)
) %>% # remove extensão
# Quebra em até 6 partes (CV, Trait, Env1, Env2, ModelRaw, Rep)
separate(
file_name,
into = c("CV", "Trait", "ModelRaw", "Rep"),
sep = "_",
fill = "right",
# preenche com NA se faltar
remove = FALSE
) %>%
# Cria coluna Env apenas se houver Env1 e Env2
mutate(
Model = sub("^Eta", "M", sub("\\.csv$", "", ModelRaw))) %>%
dplyr::select(Model, CV, Trait, Env, Rep, cor) # remove colunas auxiliares
write.csv(df, "output/Pred.ability/Pred.ability.CV1.CV2.classified.csv") # Salva consolidaçãoEste é o ponto final deste script de análise. Os kernels e outros objetos gigantes são removidos da memória, deixando apenas os dados fenotípicos brutos.
O próximo script (presumivelmente focado em visualização) irá
carregar os dois arquivos .csv limpos que acabamos de criar
(Pred.ability.CV0.CV00.csv e
Pred.ability.CV1.CV2.csv) para gerar os gráficos e tabelas
finais.
# Limpa o ambiente
rm(list = setdiff(ls(), c("Pheno", "Pedigree", "model_info")))
gc() # Limpa memória used (Mb) gc trigger (Mb) max used (Mb)
Ncells 1848468 98.8 5707711 304.9 7134638 381.1
Vcells 3617915 27.7 89972489 686.5 219659394 1675.915.5) Gráfico de barras da acurácia preditiva CV0 e CV00
Objetivo: Visualizar a acurácia preditiva média dos modelos em cada esquema de validação cruzada, destacando diferenças de desempenho.
O primeiro passo é preparar os dados para plotagem.
- Carregamos o arquivo limpo
...CV0.CV00.csv(da Seção 15.2). - Agrupamos por
Model,Env_Val,CVeTrait. - Calculamos a média (
M) e o desvio padrão (SD) da acurácia (correlação) entre as 20 repetições. - Juntamos (
left_join) com nossa tabelamodel_info(da Seção 15.5) para adicionar os nomes (Description) e tipos (Type) legíveis. - Salvamos este dataframe final e pronto para plotagem
(
...classified.csv).
# Agrupa por modelo, tipo de validação e traço
df <- read.csv("output/Pred.ability/Pred.ability.CV0.CV00.classified.csv")
df_cor <- as.data.frame(df %>%
group_by(Model, Env_Val, CV, Trait) %>%
dplyr::summarise(M = mean(cor, na.rm = TRUE), SD = sd(cor, na.rm = TRUE)))
head(df_cor)# Adiciona informações de classificação dos modelos
df_cor <- df_cor %>%
left_join(model_info, by = "Model")
head(df_cor)write.csv(df_cor, "output/Pred.ability/Pred.ability.CV0.CV00.classified.csv") # Salva consolidaçãoEste bloco cria um gráfico de exemplo para a Trait “GY”
no cenário CV “CV0”.
ggplot(...): Inicia o gráfico, mapeandoDescriptionpara o eixo X eM(acurácia média) para o eixo Y.geom_bar(): Cria as barras.geom_errorbar(): Adiciona as barras de erro (±SD).facet_wrap(. ~ Env_Val, ...): Esta é a parte importante. Ele cria um painel (um sub-gráfico) separado para cada ambiente que foi deixado de fora (ex: um painel para “CS11_WS”, um para “CS11_WW”, etc.), permitindo uma comparação fácil.
df_cor <- read.csv("output/Pred.ability/Pred.ability.CV0.CV00.classified.csv")
# Cria gráfico de barras com erro padrão e rótulos
p <- df_cor %>%
filter(CV == "CV0" & Trait == "GY") %>%
mutate(Description = reorder(Description, ModelID)) %>%
ggplot(aes(x = Description, y = M, fill = factor(Type))) +
geom_bar(stat = "identity", position = position_dodge(), width = 0.8, show.legend = FALSE) +
geom_errorbar(aes(ymin = M, ymax = M + SD),
width = .2,
position = position_dodge(.9)) +
theme_bw() +
facet_wrap(. ~ Env_Val, nrow = 4) + # Um painel para cada esquema de validação
scale_y_continuous("Prediction ability", limits = c(-0.1,1.25)) +
scale_x_discrete("Models") +
theme(axis.text.x = element_text(
angle = 90,
vjust = 0.5,
hjust = 1,
size = 6
))
# Exibe o gráfico
p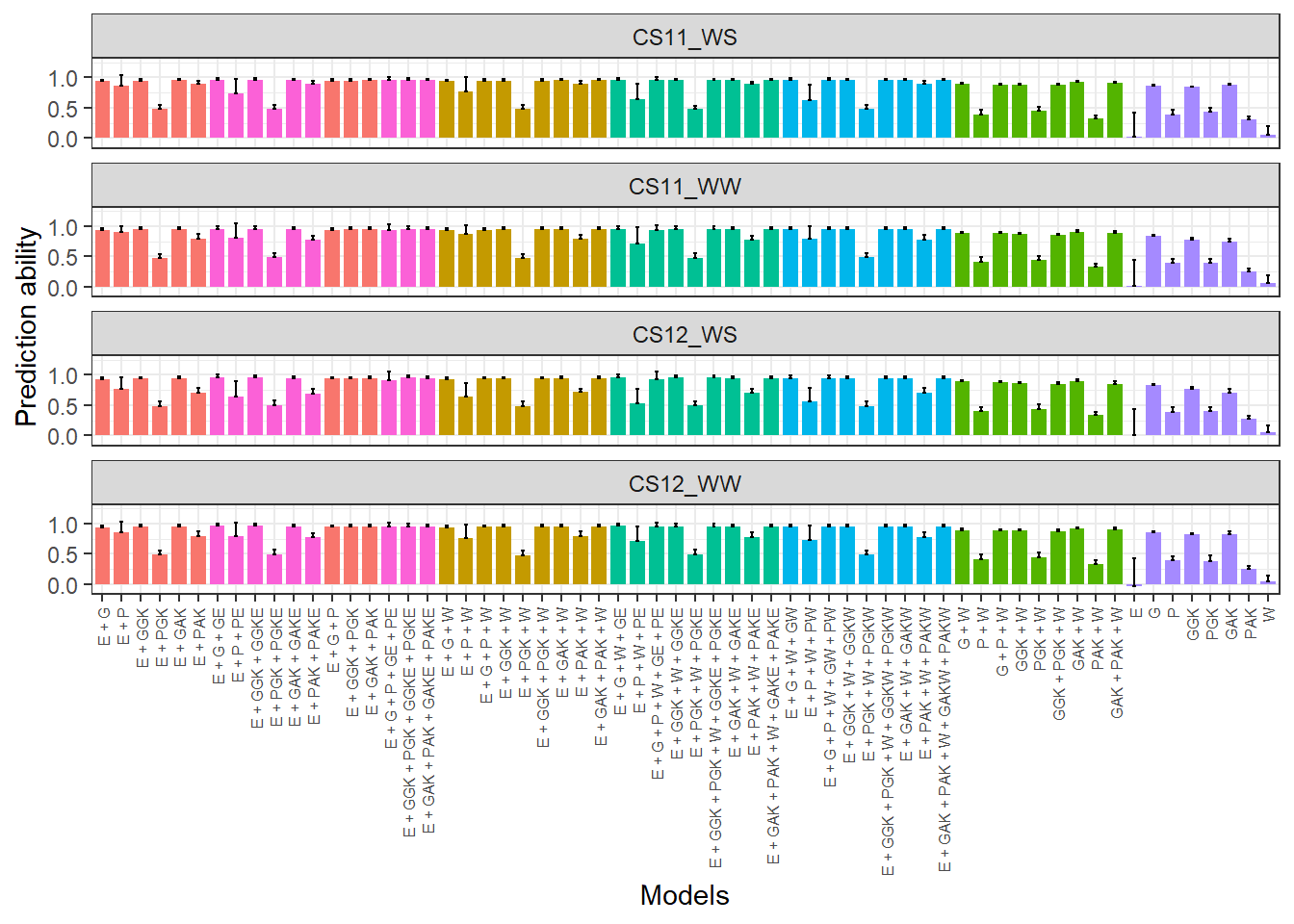
ggsave("output/Pred.ability/grafico_exemplo_GY_CV0.tiff",
plot = p,
width = 8,
height = 8, dpi = 300)df_cor <- read.csv("output/Pred.ability/Pred.ability.CV0.CV00.classified.csv")
# Cria gráfico de barras com erro padrão e rótulos
p <- df_cor %>%
group_by(Model, CV, Trait) %>%
summarise(M = mean(M, na.rm = TRUE), SD = sd(M, na.rm = TRUE)) %>%
full_join(model_info) %>%
ggplot(aes(x = reorder(Description, ModelID), y = M, fill = factor(Type))) +
geom_bar(stat = "identity", position = position_dodge(), width = 0.8, show.legend = FALSE) +
geom_errorbar(aes(ymin = M, ymax = M + SD),
width = .2,
position = position_dodge(.9)) +
theme_bw() +
facet_grid( CV ~ Trait) + # Um painel para cada esquema de validação
scale_y_continuous(limits = c(-0.1,1.25)) +
#scale_x_discrete("Models") +
theme(axis.text.x = element_text(
angle = 90,
vjust = 0.5,
hjust = 1,
size = 6
)) +
labs(x = "Models",
y = "Prediction ability")
# Exibe o gráfico
p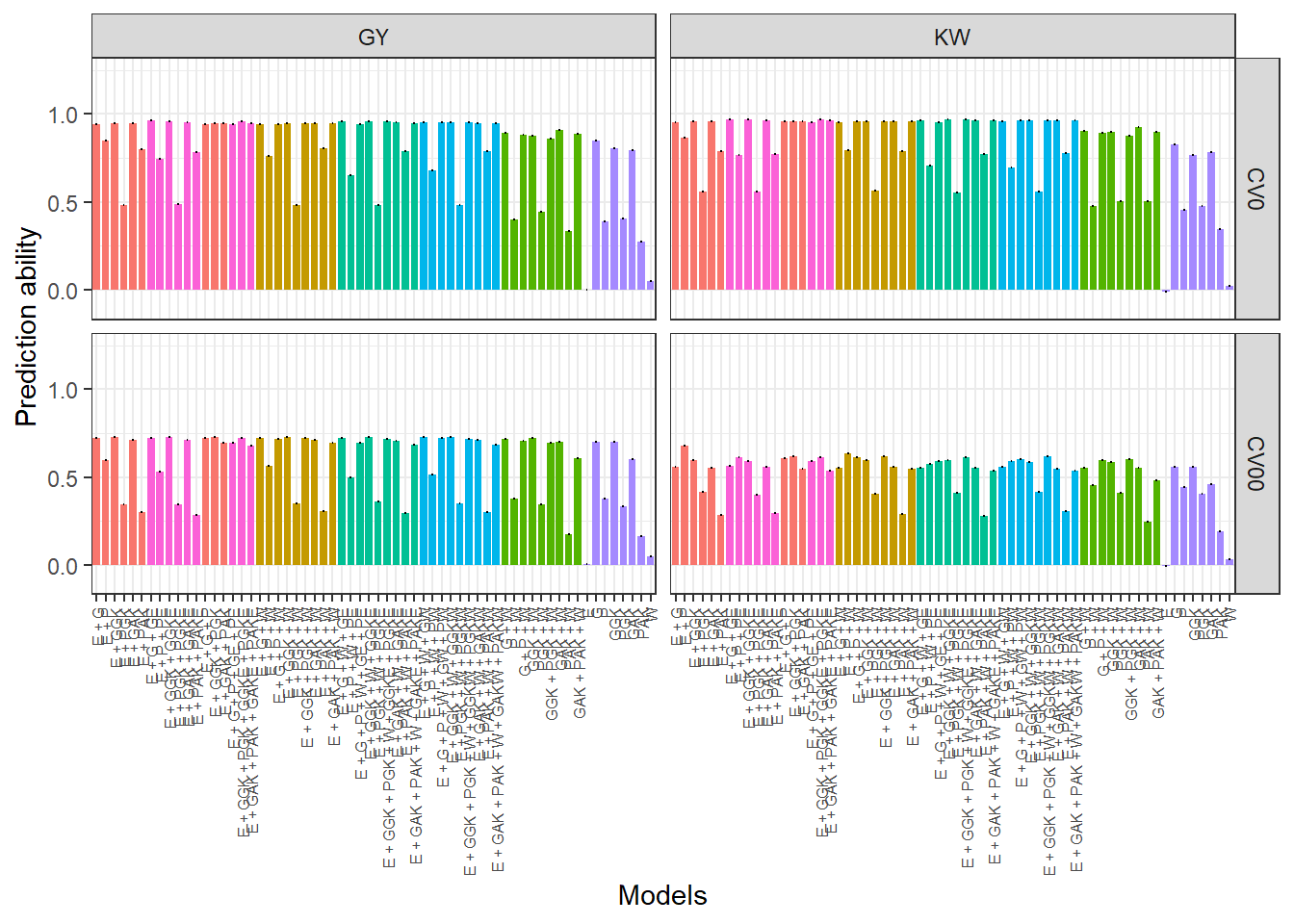
| Version | Author | Date |
|---|---|---|
| 1cbf0a0 | WevertonGomesCosta | 2025-11-03 |
ggsave("output/Pred.ability/grafico_exemplo_CV0_CV00.tiff",
plot = p,
width = 16,
height = 10, dpi = 300)15.6) Geração Automatizada de Gráficos (CV0 e CV00)
Objetivo: Automatizar a criação de gráficos para
todas as combinações de Trait, CV e
Type.
Em vez de copiar e colar o código de plotagem dezenas de vezes, este
bloco usa um loop for.
- Identificar Combinações: Primeiro, ele encontra
todas as combinações únicas de
Trait,CVeType(ex: “GY, CV0, Basico”, “GY, CV0, Clima_IntW”, “KW, CV00, Basico”, etc.). - Iterar: O loop
forpassa por cada uma dessas combinações. - Filtrar: Dentro do loop, ele filtra os
dados (
df_filtrado) apenas para a combinação atual. - Destaque Condicional:
- Calcula a acurácia média (
media_M_grupo) para aquele grupo específico. - Cria uma nova coluna
destaque. Se a acurácia de um modelo (M) for maior que a média do grupo, ele é rotulado como “Acima da Média”; caso contrário, “Abaixo/Igual à Média”.
- Calcula a acurácia média (
- Plotar:
- O
ggploté chamado, mas desta vezfill = destaqueé usado. scale_fill_manualdefine as cores (Azul para “Acima”, Cinza para “Abaixo”).facet_grid(Env_Val ~ .)cria painéis verticais (um por ambiente-deixado-de-fora), o que pode ser mais fácil de ler do que ofacet_wraphorizontal.
- O
- Salvar: O gráfico é salvo com um nome de arquivo
descritivo (ex:
grafico_GY_CV0_Basico.tiff).
Este loop irá gerar automaticamente todos os gráficos de resultados para CV0 e CV00, salvando-os no disco.
#-----------------------------------------------------------------------
# 1. CONFIGURAÇÃO INICIAL
#-----------------------------------------------------------------------
# Carregar o seu conjunto de dados
# Certifique-se de que o caminho para o arquivo está correto
df_cor <- read.csv("output/Pred.ability/Pred.ability.CV0.CV00.classified.csv") %>% as.data.frame()
# Criar um diretório para salvar os gráficos (se ele ainda não existir)
# Isso ajuda a manter os arquivos de saída organizados
output_dir <- "output/Pred.ability/graficos_gerados"
if (!dir.exists(output_dir)) {
dir.create(output_dir, recursive = TRUE)
}
#-----------------------------------------------------------------------
# 2. AUTOMAÇÃO DA GERAÇÃO DE GRÁFICOS
#-----------------------------------------------------------------------
# Identificar todas as combinações únicas das colunas de interesse
combinacoes <- df_cor %>%
distinct(Trait, CV, Env_Val, Type)
# Exibir as combinações encontradas (opcional, bom para verificação)
print("Combinações encontradas para gerar gráficos:")
head(combinacoes)
# Iniciar o loop para iterar sobre cada linha do dataframe 'combinacoes'
for (i in 1:nrow(combinacoes)) {
# Obter os valores da combinação atual
trait_atual <- combinacoes$Trait[i]
cv_atual <- combinacoes$CV[i]
type_atual <- combinacoes$Type[i]
# Mensagem de progresso no console
message(paste("Gerando gráfico para: Trait =", trait_atual, "| CV =", cv_atual, "| Type =", type_atual))
# Filtrar o dataframe principal para conter apenas os dados da combinação atual
df_filtrado <- df_cor %>%
filter(Trait == trait_atual & CV == cv_atual & Type == type_atual)
# Se não houver dados após o filtro, pular para a próxima iteração
if(nrow(df_filtrado) == 0) {
message(" -> Nenhum dado encontrado. Pulando.")
next
}
# --- ALTERAÇÃO 1: CALCULAR MÉDIA E CRIAR COLUNA DE DESTAQUE ---
# Calcular a média de 'M' *apenas* para este grupo filtrado
media_M_grupo <- mean(df_filtrado$M, na.rm = TRUE)
# Adicionar a coluna de destaque ao dataframe que será plotado
# A nova coluna 'destaque' terá um de dois valores
df_plotagem <- df_filtrado %>%
mutate(
destaque = ifelse(M > media_M_grupo,
"Acima da Média",
"Abaixo/Igual à Média")
)
# --- FIM DA ALTERAÇÃO 1 ---
# Gerar o gráfico usando o novo 'df_plotagem'
p <- df_plotagem %>%
# Ordenar Description com base em ModelID
mutate(Description = reorder(Description, ModelID)) %>%
# --- ALTERAÇÃO 2: MAPEAMENTO DO 'fill' ---
# O 'fill' (preenchimento) agora usa a nova coluna 'destaque'
ggplot(aes(x = Description, y = M, fill = destaque)) +
# 'show.legend = TRUE' (ou omitir) para mostrar a nova legenda de cores
geom_bar(stat = "identity", position = position_dodge(), show.legend = TRUE) +
geom_errorbar(aes(ymin = M, ymax = M + SD),
width = .2,
position = position_dodge(.9)) +
# Camada de texto (valores) - sem alteração
geom_text(
aes(label = round(M, 2), y = M + SD),
vjust = -0.5,
color = "black",
position = position_dodge(0.9),
angle = 0,
size = 3.5
) +
facet_grid(Env_Val ~ .) +
theme_bw() +
# Camada de escala Y (para dar espaço ao texto) - sem alteração
scale_y_continuous(
"Valor Médio (M)",
expand = expansion(mult = c(0, 0.15))
) +
# --- ALTERAÇÃO 3: DEFINIR AS CORES E LEGENDA ---
scale_fill_manual(
name = "Desempenho:", # Título da legenda
values = c("Acima da Média" = "#0072B2", # Uma cor para os valores acima (ex: Azul)
"Abaixo/Igual à Média" = "grey80") # Uma cor para os outros (ex: Cinza)
) +
labs(
title = paste("Habilidade Preditiva"),
subtitle = paste("Trait:", trait_atual, "| CV:", cv_atual, "| Type:", type_atual,
# Opcional: Adicionar a média no subtítulo
sprintf("\n(Média do grupo: %.2f)", media_M_grupo)),
x = "Descrição do Modelo"
) +
theme(
axis.text.x = element_text(
angle = 90,
vjust = 0.5,
hjust = 1,
size = 8
),
legend.position = "top" # Mover a legenda para o topo do gráfico
)
# Criar um nome de arquivo descritivo e único para o gráfico
nome_arquivo <- sprintf("%s/grafico_%s_%s_%s.tiff",
output_dir,
trait_atual,
cv_atual,
type_atual)
# Salvar o gráfico gerado no arquivo PNG
ggsave(
filename = nome_arquivo,
plot = p,
width = 10, # Largura em polegadas
height = 7, # Altura em polegadas
dpi = 300 # Resolução (qualidade da imagem)
)
}
message("\nProcesso finalizado! Gráficos salvos em '", output_dir, "'")15.7) Gráfico de barras da acurácia preditiva CV1 e CV2
Objetivo: Visualizar a acurácia preditiva média dos modelos para os cenários CV1 (predição de genótipos novos) e CV2 (predição de genótipos em ambientes novos).
O processo é idêntico ao da Seção 15.6, mas agora usamos os arquivos de resultados de CV1 e CV2.
O primeiro passo é preparar os dados para plotagem.
- Carregamos o arquivo limpo
...CV1.CV2.csv(da Seção 15.4). - Agrupamos por
Model,CVeTrait. - Calculamos a média (
M) e o desvio padrão (SD) da acurácia (correlação) entre as 20 repetições.Mserá a altura da nossa barra no gráfico, eSDserá usado para desenhar as barras de erro. - Juntamos (
left_join) com nossa tabelamodel_info(da Seção 15.5) para adicionar os nomes (Description) e tipos (Type) legíveis. - Salvamos este dataframe final e pronto para plotagem
(
...classified.csv).
# Agrupa por modelo, tipo de validação e traço
df <- read.csv("output/Pred.ability/Pred.ability.CV1.CV2.classified.csv")
df_cor <- as.data.frame(df %>%
group_by(Model, CV, Trait) %>%
dplyr::summarise(M = mean(cor, na.rm = TRUE), SD = sd(cor, na.rm = TRUE)))
head(df_cor)# Adiciona informações de classificação dos modelos
df_cor <- df_cor %>%
left_join(model_info, by = "Model")
head(df_cor)write.csv(df_cor, "output/Pred.ability/Pred.ability.CV1.CV2.classified.csv") # Salva consolidaçãoEste bloco cria um gráfico de exemplo para CV1 e CV2.
ggplot(...): Inicia o gráfico, mapeandoDescriptionpara o eixo X eM(acurácia média) para o eixo Y.geom_bar()egeom_errorbar(): Desenha as barras de acurácia e suas respectivas barras de erro (±SD).facet_grid(Trait ~ CV): Esta é a parte importante. Ele cria uma “grade” de gráficos:Trait(GY, KW) define as linhas eCV(CV1, CV2) define as colunas. Isso nos permite comparar todos os 4 cenários (GY-CV1, GY-CV2, KW-CV1, KW-CV2) em uma única figura.
df_cor <- read.csv("output/Pred.ability/Pred.ability.CV1.CV2.classified.csv")
# Cria gráfico de barras com erro padrão e rótulos
p <- df_cor %>%
filter(Trait == "GY") %>%
#mutate(Description = forcats::fct_reorder(Description, ModelID)) %>%
ggplot(aes(x = reorder(Description, ModelID), y = M, fill = factor(Type))) +
geom_bar(stat = "identity", position = position_dodge(), width = 0.8, show.legend = FALSE) +
geom_errorbar(aes(ymin = M, ymax = M + SD),
width = .2,
position = position_dodge(.9)) +
theme_bw() +
facet_grid(CV ~ .) + # Um painel para cada esquema de validação
scale_y_continuous("Prediction ability", limits = c(-0.1,1.25)) +
scale_x_discrete("Models") +
theme(axis.text.x = element_text(
angle = 90,
vjust = 0.5,
hjust = 1,
size = 6
))
# Exibe o gráfico
p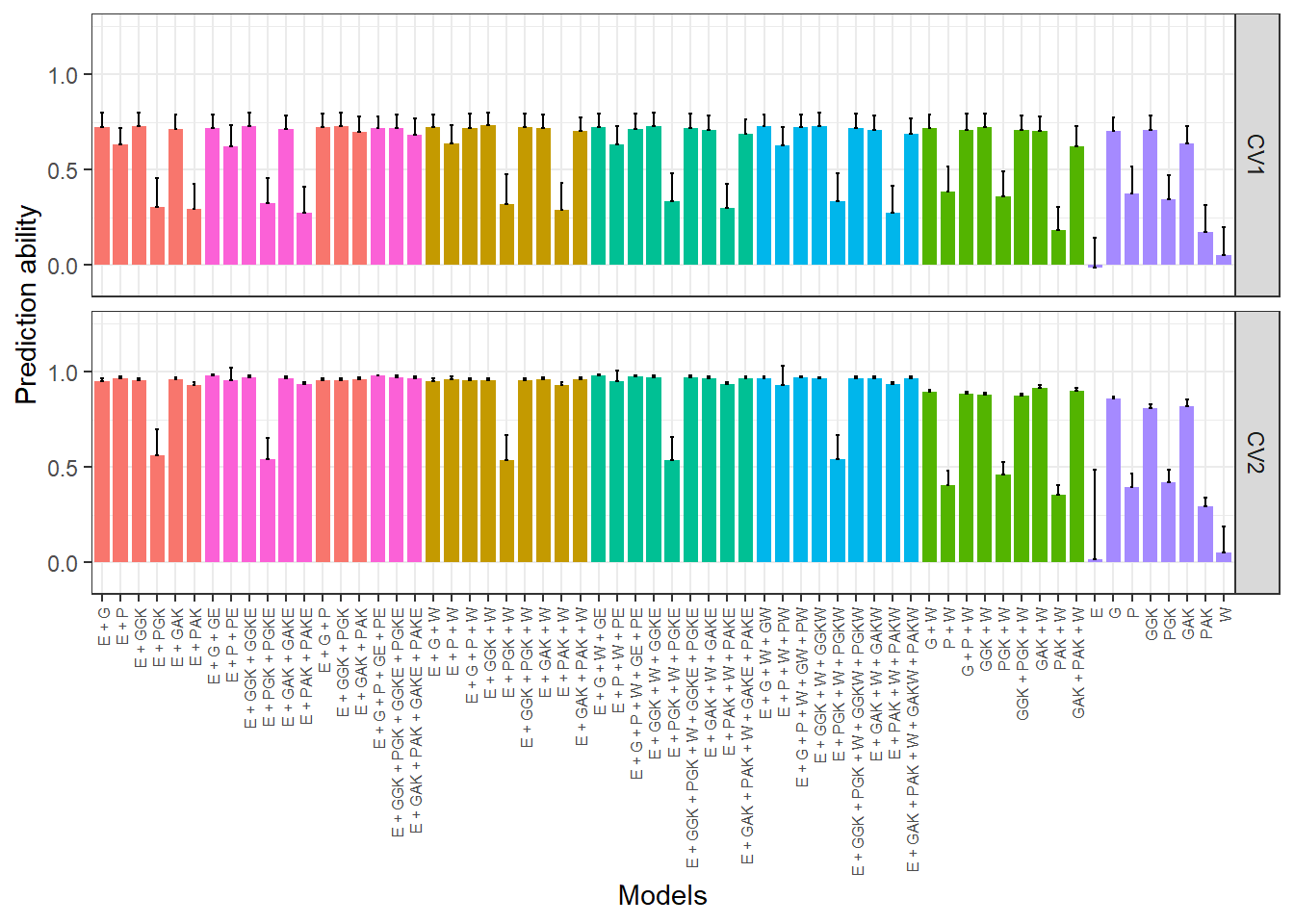
ggsave("output/Pred.ability/grafico_CV1_CV2.tiff", plot = p, width = 8, height = 8, dpi = 300)15.8) Geração Automatizada de Gráficos (CV1 e CV2)
Objetivo: Automatizar a criação de gráficos para
todas as combinações de Trait, CV e
Type.
Este loop for é idêntico ao da Seção 15.7, mas
opera sobre os dados de CV1/CV2.
- Identificar Combinações: Encontra todas as
combinações únicas de
Trait,CVeType(ex: “GY, CV1, Basico”, “GY, CV1, Interacao_E”, etc.). - Iterar: O loop
forpassa por cada uma dessas combinações. - Filtrar: Filtra os dados (
df_filtrado) apenas para a combinação atual. - Destaque Condicional:
- Calcula a acurácia média (
media_M_grupo) para aquele grupo específico. - Cria uma nova coluna
destaque(Azul para “Acima da Média”, Cinza para “Abaixo/Igual”).
- Calcula a acurácia média (
- Plotar: Cria o gráfico usando
fill = destaque. - Salvar: O gráfico é salvo com um nome de arquivo
descritivo (ex:
grafico_GY_CV1_Basico.tiff).
Este loop irá gerar automaticamente todos os gráficos de resultados para CV1 e CV2, salvando-os no disco.
#-----------------------------------------------------------------------
# 1. CONFIGURAÇÃO INICIAL
#-----------------------------------------------------------------------
# Carregar o seu conjunto de dados
# Certifique-se de que o caminho para o arquivo está correto
df_cor <- read.csv("output/Pred.ability/Pred.ability.CV1.CV2.classified.csv")
# Criar um diretório para salvar os gráficos (se ele ainda não existir)
# Isso ajuda a manter os arquivos de saída organizados
output_dir <- "output/Pred.ability/graficos_gerados"
if (!dir.exists(output_dir)) {
dir.create(output_dir, recursive = TRUE)
}
#-----------------------------------------------------------------------
# 2. AUTOMAÇÃO DA GERAÇÃO DE GRÁFICOS
#-----------------------------------------------------------------------
# Identificar todas as combinações únicas das colunas de interesse
combinacoes <- df_cor %>%
distinct(Trait, CV, Type)
# Exibir as combinações encontradas (opcional, bom para verificação)
print("Combinações encontradas para gerar gráficos:")
print(combinacoes)
# Iniciar o loop para iterar sobre cada linha do dataframe 'combinacoes'
for (i in 1:nrow(combinacoes)) {
# Obter os valores da combinação atual
trait_atual <- combinacoes$Trait[i]
cv_atual <- combinacoes$CV[i]
type_atual <- combinacoes$Type[i]
# Mensagem de progresso no console
message(paste("Gerando gráfico para: Trait =", trait_atual, "| CV =", cv_atual, "| Type =", type_atual))
# Filtrar o dataframe principal para conter apenas os dados da combinação atual
df_filtrado <- df_cor %>%
filter(Trait == trait_atual & CV == cv_atual & Type == type_atual)
# Se não houver dados após o filtro, pular para a próxima iteração
if(nrow(df_filtrado) == 0) {
message(" -> Nenhum dado encontrado. Pulando.")
next
}
# --- ALTERAÇÃO 1: CALCULAR MÉDIA E CRIAR COLUNA DE DESTAQUE ---
# Calcular a média de 'M' *apenas* para este grupo filtrado
media_M_grupo <- mean(df_filtrado$M, na.rm = TRUE)
# Adicionar a coluna de destaque ao dataframe que será plotado
# A nova coluna 'destaque' terá um de dois valores
df_plotagem <- df_filtrado %>%
mutate(
destaque = ifelse(M > media_M_grupo,
"Acima da Média",
"Abaixo/Igual à Média")
)
# --- FIM DA ALTERAÇÃO 1 ---
# Gerar o gráfico usando o novo 'df_plotagem'
p <- df_plotagem %>%
# Ordenar Description com base em ModelID
mutate(Description = reorder(Description, ModelID)) %>%
# --- ALTERAÇÃO 2: MAPEAMENTO DO 'fill' ---
# O 'fill' (preenchimento) agora usa a nova coluna 'destaque'
ggplot(aes(x = Description, y = M, fill = destaque)) +
# 'show.legend = TRUE' (ou omitir) para mostrar a nova legenda de cores
geom_bar(stat = "identity", position = position_dodge(), show.legend = TRUE) +
geom_errorbar(aes(ymin = M, ymax = M + SD),
width = .2,
position = position_dodge(.9)) +
# Camada de texto (valores) - sem alteração
geom_text(
aes(label = round(M, 2), y = M + SD),
vjust = -0.5,
color = "black",
position = position_dodge(0.9),
angle = 0,
size = 3.5
) +
theme_bw() +
# Camada de escala Y (para dar espaço ao texto) - sem alteração
scale_y_continuous(
"Valor Médio (M)",
expand = expansion(mult = c(0, 0.15))
) +
# --- ALTERAÇÃO 3: DEFINIR AS CORES E LEGENDA ---
scale_fill_manual(
name = "Desempenho:", # Título da legenda
values = c("Acima da Média" = "#0072B2", # Uma cor para os valores acima (ex: Azul)
"Abaixo/Igual à Média" = "grey80") # Uma cor para os outros (ex: Cinza)
) +
labs(
title = paste("Habilidade Preditiva"),
subtitle = paste("Trait:", trait_atual, "| CV:", cv_atual, "| Type:", type_atual,
# Opcional: Adicionar a média no subtítulo
sprintf("\n(Média do grupo: %.2f)", media_M_grupo)),
x = "Descrição do Modelo"
) +
theme(
axis.text.x = element_text(
angle = 90,
vjust = 0.5,
hjust = 1,
size = 8
),
legend.position = "top" # Mover a legenda para o topo do gráfico
)
# Criar um nome de arquivo descritivo e único para o gráfico
nome_arquivo <- sprintf("%s/grafico_%s_%s_%s.tiff",
output_dir,
trait_atual,
cv_atual,
type_atual)
# Salvar o gráfico gerado no arquivo PNG
ggsave(
filename = nome_arquivo,
plot = p,
width = 10, # Largura em polegadas
height = 7, # Altura em polegadas
dpi = 300 # Resolução (qualidade da imagem)
)
}
message("\nProcesso finalizado! Gráficos salvos em '", output_dir, "'")
sessionInfo()R version 4.5.1 (2025-06-13 ucrt)
Platform: x86_64-w64-mingw32/x64
Running under: Windows 11 x64 (build 26200)
Matrix products: default
LAPACK version 3.12.1
locale:
[1] LC_COLLATE=Portuguese_Brazil.utf8 LC_CTYPE=Portuguese_Brazil.utf8
[3] LC_MONETARY=Portuguese_Brazil.utf8 LC_NUMERIC=C
[5] LC_TIME=Portuguese_Brazil.utf8
time zone: America/Sao_Paulo
tzcode source: internal
attached base packages:
[1] grid parallel stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] gridExtra_2.3 MASS_7.3-65 doParallel_1.0.17 iterators_1.0.14
[5] foreach_1.5.2 BGLR_1.1.4 data.table_1.17.8 lubridate_1.9.4
[9] forcats_1.0.0 stringr_1.5.2 dplyr_1.1.4 purrr_1.1.0
[13] readr_2.1.5 tidyr_1.3.1 tibble_3.3.0 ggplot2_4.0.0
[17] tidyverse_2.0.0
loaded via a namespace (and not attached):
[1] sass_0.4.10 generics_0.1.4 stringi_1.8.7 hms_1.1.3
[5] digest_0.6.37 magrittr_2.0.4 evaluate_1.0.5 timechange_0.3.0
[9] RColorBrewer_1.1-3 fastmap_1.2.0 rprojroot_2.1.1 workflowr_1.7.2
[13] jsonlite_2.0.0 whisker_0.4.1 promises_1.3.3 scales_1.4.0
[17] truncnorm_1.0-9 textshaping_1.0.3 codetools_0.2-20 jquerylib_0.1.4
[21] cli_3.6.5 rlang_1.1.6 withr_3.0.2 cachem_1.1.0
[25] yaml_2.3.10 tools_4.5.1 tzdb_0.5.0 httpuv_1.6.16
[29] vctrs_0.6.5 R6_2.6.1 lifecycle_1.0.4 git2r_0.36.2
[33] fs_1.6.6 ragg_1.5.0 pkgconfig_2.0.3 pillar_1.11.1
[37] bslib_0.9.0 later_1.4.4 gtable_0.3.6 glue_1.8.0
[41] Rcpp_1.1.0 systemfonts_1.2.3 xfun_0.53 tidyselect_1.2.1
[45] rstudioapi_0.17.1 knitr_1.50 farver_2.1.2 htmltools_0.5.8.1
[49] labeling_0.4.3 rmarkdown_2.29 compiler_4.5.1 S7_0.2.0