Simulation
Zheng Li
2022-02-06
Last updated: 2022-03-10
Checks: 7 0
Knit directory: BASS-analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(0) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version b835db9. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
working directory clean
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/simu.Rmd) and HTML (docs/simu.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | b835db9 | zhengli09 | 2022-03-10 | Add figure of additional tissue sections |
| html | 182beeb | zhengli09 | 2022-03-10 | Build site. |
| Rmd | 34e84d6 | zhengli09 | 2022-03-10 | Add software tutorial and multi-sample simulation |
| html | a27dd42 | zhengli09 | 2022-02-27 | Build site. |
| Rmd | ada7b84 | zhengli09 | 2022-02-27 | Modify links |
| html | 9a68511 | zhengli09 | 2022-02-27 | Build site. |
| Rmd | 4ec8df1 | zhengli09 | 2022-02-27 | Publish the introduction, simulation analysis and STARmap analysis |
Introduction
This page provides code to reproduce all the simulation results for BASS. Hopefully, this can also serve as a resource for building up your simulation studies and evaluate your statistical methods.
Simulation study
Generate simulated data
The simulation study relies on a few R packages and python softwares (e.g. SpaGCN and FICT), which need to be installed beforehand. The specific packages and all necessary functions to conduct the simulation study can be found at simulation.
source("code/simu_utils.R")
# data from STARmap (BZ5) for inferring parameters for splatter
cnts <- readRDS("data/simu_cnts.RDS")
info <- readRDS("data/simu_info.RDS")
xy <- as.matrix(info[, c("x", "y")])
starmap <- list(cnts = cnts, info = info)
# we perform 50 simulation replicates for each setting
# The random seeds used for each replicate are generated as below
NREP <- 50
set.seed(0)
seeds <- sample(20201230, NREP)We generate the simulated data under the baseline simulation setting. Refer to the params file for a complete list of simulation settings.
# baseline settings for all four scenarios for the
# single tissue section analysis (L = 1)
scenarios <- 1:4 # simulation scenarios
C <- 4 # number of cell types
R <- 4 # number of spatial domains
J <- 200 # number of genes
de_prop <- 0.2 # proportion of DE genes for each cell type
de_facLoc <- 1.1 # DE gene strength
L <- 1 # number of tissue sections
# illustrate the data at the first replicate
rep <- 1
sim_dats <- lapply(scenarios, function(scenario){
simu(starmap = starmap, scenario = scenario, C = C, J = J, L = L,
batch_facLoc = 0, de_prop = de_prop, de_facLoc = de_facLoc,
de_facScale = 0.4, sim_seed = seeds[rep], debug = FALSE)
})# The simulated data include a list of L gene expression count matrices,
# a list of L true cell type label vectors for evaluation purpose, and
# the random seed used for generating the data
scenario <- 3
sim_dat <- sim_dats[[scenario]]
# gene expression count matrix of the first tissue section under scenario 3
sim_dat[[1]][[1]][1:5, 1:5] Cell1 Cell2 Cell3 Cell4 Cell5
Gene1 0 0 0 0 0
Gene2 17 2 6 9 1
Gene3 0 0 0 0 0
Gene4 0 0 0 0 19
Gene5 0 1 3 0 44Visualize of the simulated data
We visualize the cell type distributions under scenarios I-IV. You can refer to visualization for some useful plotting functions or you can write your own code for plotting.
library(cowplot)
source("code/viz.R")
p <- lapply(scenarios, function(scenario){
plotClusters(xy, labels = sim_dats[[scenario]][[2]][[1]],
title = paste("Scenario", c("I", "II", "III", "IV")[scenario]))
})
legend <- get_legend(p[[1]] +
theme(legend.position = "bottom") +
guides(color = guide_legend(title = "Cell type")))
p <- plot_grid(plotlist = p, ncol = 2)
plot_grid(p, legend, ncol = 1, rel_heights = c(1, 0.1))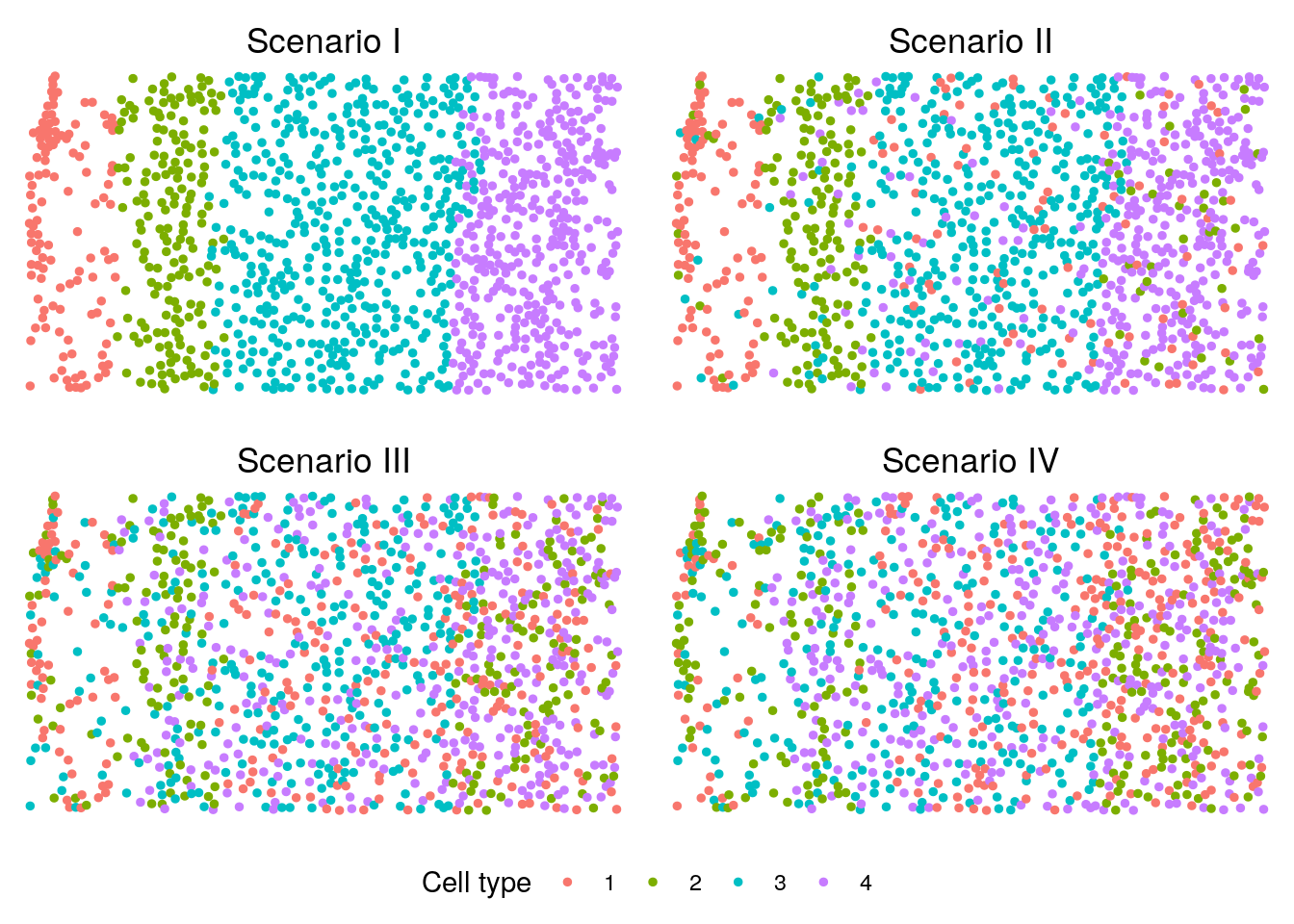
Running all methods
We have wrapped up code for running different methods into different functions. Those functions take in simulated data and output adjusted random index (ARI) measuring the estimate cell type or spatial domain labels with the underlying truth. Functions for running different methods can be found at simulation. We take the simulated data from scenario 3 as an example to illustrate running different methods.
scenario <- 3Run BASS
BASS_out <- run_BASS(sim_dats[[scenario]], xy, "ACCUR_EST", beta = 1, C, R,
init_method = "kmeans", cov_struc = "EEE")***************************************
INPUT INFO:
- Number of tissue sections: 1
- Number of cells/spots: 1127
- Number of genes: 200
- Potts interaction parameter estimation approach: ACCUR_EST
- Variance-covariance structure of gene expression features: EEE
To list all hyper-parameters, Type listAllHyper(BASS_object)
***************************************
***** Log-normalize gene expression data *****
***** Exclude genes with 0 expression *****
***** Reduce data dimension with PCA *****
......................................................................................
. Method Time (sec) Status .
......................................................................................
. ECR-ITERATIVE-1 15.013 Converged (2 iterations) .
......................................................................................
Relabelling all methods according to method ECR-ITERATIVE-1 ... done!
Retrieve the 1 permutation arrays by typing:
[...]$permutations$"ECR-ITERATIVE-1"
Retrieve the 1 best clusterings: [...]$clusters
Retrieve the 1 CPU times: [...]$timings
Retrieve the 1 X 1 similarity matrix: [...]$similarity
Label switching finished. Total time: 17.1 seconds.
......................................................................................
. Method Time (sec) Status .
......................................................................................
. ECR-ITERATIVE-1 12.159 Converged (2 iterations) .
......................................................................................
Relabelling all methods according to method ECR-ITERATIVE-1 ... done!
Retrieve the 1 permutation arrays by typing:
[...]$permutations$"ECR-ITERATIVE-1"
Retrieve the 1 best clusterings: [...]$clusters
Retrieve the 1 CPU times: [...]$timings
Retrieve the 1 X 1 similarity matrix: [...]$similarity
Label switching finished. Total time: 13.2 seconds. BASS_out$c_ari
[1] 0.9670117
$z_ari
[1] 0.9196672
$pi_est
[,1] [,2] [,3] [,4]
[1,] 0.5241935 0.005882353 0.2652632 0.2458101
[2,] 0.1935484 0.488235294 0.0000000 0.2569832
[3,] 0.2822581 0.247058824 0.4863158 0.0000000
[4,] 0.0000000 0.258823529 0.2484211 0.4972067
$mse_pi
[1] 0.04606247Run HMRF
Please note that running HMRF requires you to specify a path for python and will generate intermediate and final results into a specified folder. Please modify those directories accordingly in the run_HMRF function if you want to use the function for your own analysis.
# Run HMRF
# Note that case and rep parameters below are used to generate a unique
# temporary directories for running HMRF.
HMRF_out <- run_HMRF(sim_dat, xy, ztrue = info$z, R, case = 3,
rep, usePCs = F, dosearchSEgenes = T)
external python path provided and will be used
Consider to install these (optional) packages to run all possible Giotto commands for spatial analyses: trendsceek multinet RTriangle FactoMiner
Giotto does not automatically install all these packages as they are not absolutely required and this reduces the number of dependencies
This is the single parameter version of binSpect
1. matrix binarization complete
2. spatial enrichment test completed
3. (optional) average expression of high expressing cells calculated
4. (optional) number of high expressing cells calculated
[1] "/net/mulan/home/zlisph//softwares/miniconda3/bin/python /net/mulan/home/zlisph/R/x86_64-pc-linux-gnu-library/4.0/Giotto/python/reader2.py -l \"/net/mulan/home/zlisph/BASS_pjt/2_simu/HMRF_tmp/case3/rep1/result_k4/spatial_cell_locations.txt\" -g \"/net/mulan/home/zlisph/BASS_pjt/2_simu/HMRF_tmp/case3/rep1/result_k4/spatial_genes.txt\" -n \"/net/mulan/home/zlisph/BASS_pjt/2_simu/HMRF_tmp/case3/rep1/result_k4/spatial_network.txt\" -e \"/net/mulan/home/zlisph/BASS_pjt/2_simu/HMRF_tmp/case3/rep1/result_k4/expression_matrix.txt\" -o \"/net/mulan/home/zlisph/BASS_pjt/2_simu/HMRF_tmp/case3/rep1/result_k4/result.spatial.zscore\" -a test -k 4 -b 0 2 26 -t 1e-10 -z none -s 100 -i 100"# ARI for spatial domain detection across different
# spatial interaction parameter betas.
HMRF_out 0.0 2.0 4.0 6.0 8.0 10.0 12.0 14.0
0.1368218 0.1398503 0.1466260 0.1940655 0.2686491 0.3115478 0.3773713 0.3902822
16.0 18.0 20.0 22.0 24.0 26.0 28.0 30.0
0.4737238 0.4873337 0.4892104 0.5113665 0.5009481 0.5214409 0.5543050 0.5585267
32.0 34.0 36.0 38.0 40.0 42.0 44.0 46.0
0.5459983 0.5794579 0.5894143 0.5809235 0.5182257 0.5254769 0.5411271 0.5335083
48.0 50.0
0.5187454 0.5167944 Run BayesSpace
Note that BayesSpace cannot identify any neighbors because it was developed for analyzing ordered lattice structure of spots from ST and 10x Visium platforms.
# Run BayesSpace
BayesSpace_out <- run_BayesSpace(sim_dat, xy, ztrue = info$z, R)Neighbors were identified for 0 out of 1127 spots.Fitting model...Calculating labels using iterations 1000 through 10000.# ARI for spatial domain detection
BayesSpace_out[1] 0.1515157Run SpaGCN
Because SpaGCN was originally developed with Python, we wrapped up the code into a function and have it imported into R such that we can conveniently analyze the simulated data in R. The python function can be found at simulation.
# Run SpaGCN
source_python("code/run_SpaGCN.py")
SpaGCN_out <- run_SpaGCN(sim_dat, xy, info$z, R)
# ARI for spatial domain detection
SpaGCN_out[1] 0.2346377Run Seurat
Because Seurat uses a resolution parameter to indirectly determine the number of clusters, we run Seurat on a range of resolution parameters and identify the first value that resulted in the desired number of cell types.
# Run Seurat
resolutions <- seq(0.3, 1.5, by = 0.1)
seu_out <- seu_cluster(sim_dat, resolutions)
# ARI for cell type clustering and the number of identified clusters
seu_out [1] "0.930823007047579,4" "0.933410743550603,4" "0.933410743550603,4"
[4] "0.9355780298241,4" "0.9355780298241,4" "0.9355780298241,4"
[7] "0.9355780298241,4" "0.9355780298241,4" "0.820473068330246,5"
[10] "0.818347732690449,5" "0.818347732690449,5" "0.818097321908858,6"
[13] "0.815690823967137,6"Run SC3
Note that when the number of cells exceeds 5,000, SC3 randomly selects 5,000 cells for clustering, trains a support vector machine with the cluster labels obtained for the 5,000 cells, and then predict the cluster labels of the remaining cells.
# Run SC3
# Note SC3 algorithm is slow
sc3_out <- sc3_cluster(sim_dat, C)Setting SC3 parameters...Calculating distances between the cells...Performing transformations and calculating eigenvectors...Performing k-means clustering...Calculating consensus matrix...# ARI for cell type clustering
sc3_out[1] 0.9602983Run FICT
Please install the software according to the FICT github and modify the paths to the FICT software and a temporary folder accordingly in the fict_cluster function.
# Note that case and rep parameters below are used to generate a unique
# temporary directories for running FICT.
fict_out <- fict_cluster(sim_dat, xy, C, case = 3, rep)
# ARI for cell type clustering
fict_out[1] 0.9566095Multi-sample simulation
In addition to simulating and analyzing a single tissue section, we also simulated multiple tissue sections and evaluated the performance of BASS for multi-sample integrative analysis. Specifically, we generated additional tissue sections based on the same spatial locations of the 1,127 cells in the single tissue section analysis but with slightly different spatial domain allocations for them, creating varying spatial domain boundaries between the four cortical layers on different tissue sections. In each section, we set the spatial domain boundaries to be vertical, and created new boundaries by horizontally moving the original boundaries based on a uniform distribution with step size set to be approximately 10% (= 500) of the tissue width. The resulting new tissue sections are as follows. 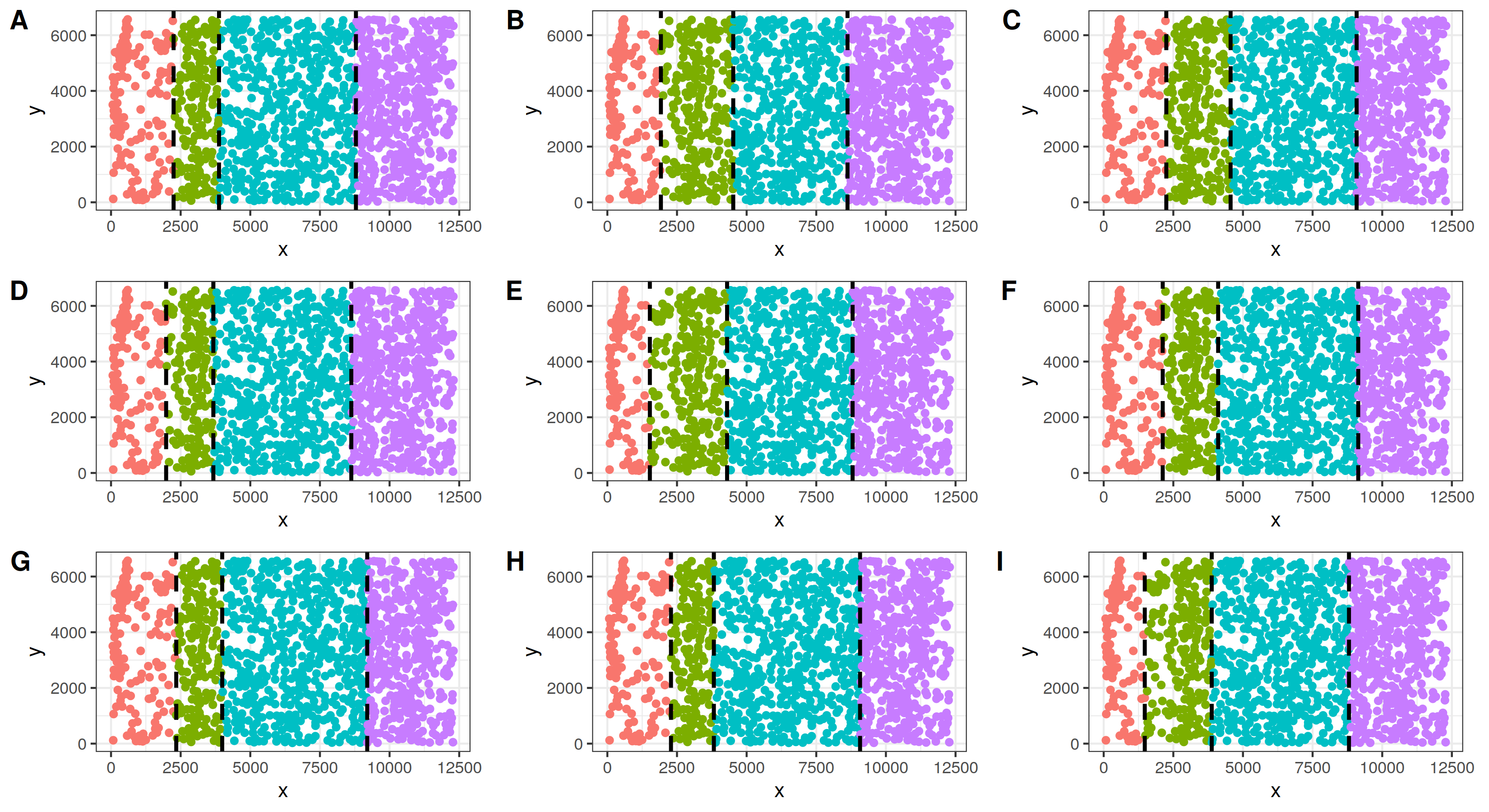
Here, similar to the single-sample analysis, we generate the simulated data under the baseline simulation setting but for five tissue sections (L = 5). Refer to the params_mult file for a complete list of simulation settings.
# generate simulated data for five tissue sections
L <- 5
sim_dat <- simu(
starmap = starmap, scenario = scenario, C = C, J = J, L = L,
batch_facLoc = 0, de_prop = de_prop, de_facLoc = de_facLoc,
de_facScale = 0.4, sim_seed = seeds[rep], debug = FALSE)
# run BASS
BASS_out <- run_BASS(sim_dat, xy, "ACCUR_EST", beta = 1, C, R,
init_method = "kmeans", cov_struc = "EEE")***************************************
INPUT INFO:
- Number of tissue sections: 5
- Number of cells/spots: 1127 1127 1127 1127 1127
- Number of genes: 200
- Potts interaction parameter estimation approach: ACCUR_EST
- Variance-covariance structure of gene expression features: EEE
To list all hyper-parameters, Type listAllHyper(BASS_object)
***************************************
***** Log-normalize gene expression data *****
***** Exclude genes with 0 expression *****
***** Reduce data dimension with PCA *****
***** Correct batch effect with Harmony *****
......................................................................................
. Method Time (sec) Status .
......................................................................................
. ECR-ITERATIVE-1 72.63 Converged (3 iterations) .
......................................................................................
Relabelling all methods according to method ECR-ITERATIVE-1 ... done!
Retrieve the 1 permutation arrays by typing:
[...]$permutations$"ECR-ITERATIVE-1"
Retrieve the 1 best clusterings: [...]$clusters
Retrieve the 1 CPU times: [...]$timings
Retrieve the 1 X 1 similarity matrix: [...]$similarity
Label switching finished. Total time: 80 seconds.
......................................................................................
. Method Time (sec) Status .
......................................................................................
. ECR-ITERATIVE-1 46.069 Converged (2 iterations) .
......................................................................................
Relabelling all methods according to method ECR-ITERATIVE-1 ... done!
Retrieve the 1 permutation arrays by typing:
[...]$permutations$"ECR-ITERATIVE-1"
Retrieve the 1 best clusterings: [...]$clusters
Retrieve the 1 CPU times: [...]$timings
Retrieve the 1 X 1 similarity matrix: [...]$similarity
Label switching finished. Total time: 52.7 seconds. BASS_out$c_ari
[1] 0.9639116
$z_ari
[1] 0.9295722
$pi_est
[,1] [,2] [,3] [,4]
[1,] 0.51017214 0.0000000 0.2521367521 0.2521091811
[2,] 0.27073552 0.5028571 0.0004748338 0.2461538462
[3,] 0.21596244 0.2331429 0.4971509972 0.0004962779
[4,] 0.00312989 0.2640000 0.2502374169 0.5012406948
$mse_pi
[1] 0.01290559Find all the simulation results
Put the link to all the simulation results here later and plot the two main figures on simulation analysis.
sessionInfo()R version 4.1.2 (2021-11-01)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 18.04.6 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/libopenblasp-r0.2.20.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] cowplot_1.1.1 reticulate_1.22
[3] splatter_1.16.1 gtools_3.9.2
[5] scater_1.16.2 forcats_0.5.0
[7] stringr_1.4.0 dplyr_1.0.8
[9] purrr_0.3.4 readr_1.3.1
[11] tidyr_1.1.1 tibble_3.1.6
[13] ggplot2_3.3.5 tidyverse_1.3.0
[15] SC3_1.20.0 Seurat_3.2.3
[17] BayesSpace_1.2.1 SingleCellExperiment_1.14.1
[19] SummarizedExperiment_1.22.0 Biobase_2.48.0
[21] GenomicRanges_1.44.0 GenomeInfoDb_1.24.2
[23] IRanges_2.26.0 S4Vectors_0.30.2
[25] BiocGenerics_0.38.0 MatrixGenerics_1.4.3
[27] matrixStats_0.61.0 Giotto_1.0.4
[29] BASS_1.0 mclust_5.4.9
[31] GIGrvg_0.5 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] rappdirs_0.3.3 scattermore_0.7
[3] maxLik_1.5-2 coda_0.19-4
[5] bit64_4.0.2 knitr_1.37
[7] irlba_2.3.3 DelayedArray_0.18.0
[9] data.table_1.14.2 rpart_4.1-15
[11] RCurl_1.98-1.5 doParallel_1.0.15
[13] generics_0.1.2 callr_3.7.0
[15] RSQLite_2.2.0 combinat_0.0-8
[17] RANN_2.6.1 proxy_0.4-26
[19] future_1.22.1 bit_4.0.4
[21] xml2_1.3.3 lubridate_1.7.9
[23] spatstat.data_2.1-0 httpuv_1.5.4
[25] assertthat_0.2.1 viridis_0.5.1
[27] xfun_0.29 hms_0.5.3
[29] jquerylib_0.1.4 evaluate_0.15
[31] promises_1.1.1 DEoptimR_1.0-9
[33] fansi_1.0.2 readxl_1.3.1
[35] dbplyr_1.4.4 igraph_1.2.7
[37] DBI_1.1.1 htmlwidgets_1.5.1
[39] ellipsis_0.3.2 backports_1.2.1
[41] deldir_1.0-6 sparseMatrixStats_1.4.2
[43] vctrs_0.3.8 ROCR_1.0-11
[45] abind_1.4-5 withr_2.4.3
[47] cachem_1.0.6 robustbase_0.93-9
[49] checkmate_2.0.0 sctransform_0.3.2
[51] scran_1.20.1 goftest_1.2-3
[53] cluster_2.1.2 lazyeval_0.2.2
[55] crayon_1.5.0 labeling_0.4.2
[57] edgeR_3.34.1 pkgconfig_2.0.3
[59] nlme_3.1-153 vipor_0.4.5
[61] rlang_1.0.1 globals_0.14.0
[63] lifecycle_1.0.1 miniUI_0.1.1.1
[65] sandwich_3.0-1 BiocFileCache_1.12.0
[67] modelr_0.1.8 rsvd_1.0.3
[69] cellranger_1.1.0 rprojroot_2.0.2
[71] polyclip_1.10-0 lmtest_0.9-38
[73] rngtools_1.5.2 Matrix_1.3-4
[75] Rhdf5lib_1.10.1 zoo_1.8-9
[77] reprex_0.3.0 beeswarm_0.4.0
[79] whisker_0.4 ggridges_0.5.3
[81] processx_3.5.2 pheatmap_1.0.12
[83] png_0.1-7 viridisLite_0.4.0
[85] bitops_1.0-7 getPass_0.2-2
[87] KernSmooth_2.23-20 blob_1.2.1
[89] DelayedMatrixStats_1.14.3 doRNG_1.8.2
[91] parallelly_1.28.1 lpSolve_5.6.15
[93] beachmat_2.8.1 scales_1.1.1
[95] memoise_2.0.1 magrittr_2.0.2
[97] plyr_1.8.6 ica_1.0-2
[99] zlibbioc_1.34.0 DirichletReg_0.7-1
[101] compiler_4.1.2 miscTools_0.6-26
[103] dqrng_0.3.0 RColorBrewer_1.1-2
[105] rrcov_1.6-0 fitdistrplus_1.1-6
[107] cli_3.2.0 XVector_0.32.0
[109] listenv_0.8.0 patchwork_1.1.1
[111] pbapply_1.5-0 ps_1.6.0
[113] Formula_1.2-4 MASS_7.3-54
[115] mgcv_1.8-38 tidyselect_1.1.2
[117] stringi_1.7.6 highr_0.9
[119] yaml_2.3.5 BiocSingular_1.4.0
[121] locfit_1.5-9.4 ggrepel_0.9.1
[123] grid_4.1.2 sass_0.4.0
[125] tools_4.1.2 future.apply_1.8.1
[127] label.switching_1.8 rstudioapi_0.13
[129] bluster_1.2.1 foreach_1.5.0
[131] git2r_0.28.0 metapod_1.0.0
[133] gridExtra_2.3 farver_2.1.0
[135] Rtsne_0.15 digest_0.6.29
[137] pracma_2.2.9 shiny_1.5.0
[139] Rcpp_1.0.8 broom_0.7.10
[141] scuttle_1.2.1 harmony_0.1.0
[143] later_1.1.0.1 RcppAnnoy_0.0.19
[145] WriteXLS_6.3.0 httr_1.4.2
[147] Rdpack_2.1.2 colorspace_2.0-3
[149] rvest_0.3.6 fs_1.5.2
[151] tensor_1.5 splines_4.1.2
[153] uwot_0.1.10 statmod_1.4.36
[155] spatstat.utils_2.2-0 xgboost_1.4.1.1
[157] plotly_4.9.2.1 xtable_1.8-4
[159] jsonlite_1.8.0 spatstat_1.64-1
[161] R6_2.5.1 pillar_1.7.0
[163] htmltools_0.5.2 mime_0.12
[165] glue_1.6.2 fastmap_1.1.0
[167] BiocParallel_1.22.0 BiocNeighbors_1.6.0
[169] class_7.3-19 codetools_0.2-18
[171] pcaPP_1.9-74 mvtnorm_1.1-3
[173] utf8_1.2.2 lattice_0.20-45
[175] bslib_0.3.1 curl_4.3.2
[177] ggbeeswarm_0.6.0 leiden_0.3.9
[179] survival_3.2-13 limma_3.48.3
[181] rmarkdown_2.12.1 munsell_0.5.0
[183] e1071_1.7-9 rhdf5_2.32.2
[185] GenomeInfoDbData_1.2.6 iterators_1.0.13
[187] smfishHmrf_0.1 haven_2.3.1
[189] reshape2_1.4.4 gtable_0.3.0
[191] rbibutils_2.2.4