05 cytotoxic T cell quantification
Daniel
July 2020
Last updated: 2022-02-25
Checks: 6 1
Knit directory: MelanomaIMC/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200728) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 0deb8ee. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rproj.user/
Ignored: code/.DS_Store
Ignored: code/._.DS_Store
Ignored: data/.DS_Store
Ignored: data/._.DS_Store
Ignored: data/data_for_analysis/
Ignored: data/data_for_analysis_zenodo/
Ignored: data/full_data.zip
Ignored: data/full_data/
Unstaged changes:
Modified: .gitignore
Modified: analysis/09_Tcell_Score.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/09_Tcell_Score.Rmd) and HTML (docs/09_Tcell_Score.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | fe331cb | toobiwankenobi | 2022-02-22 | re-run whole analysis |
| html | 73aa800 | toobiwankenobi | 2022-02-22 | add .html for static website |
| Rmd | 3da15db | toobiwankenobi | 2021-11-24 | changes for revision |
| html | 4109ff1 | toobiwankenobi | 2021-07-07 | delete html files and adapt gitignore |
| Rmd | 3203891 | toobiwankenobi | 2021-02-19 | change celltype names |
| html | 3203891 | toobiwankenobi | 2021-02-19 | change celltype names |
| Rmd | ee1595d | toobiwankenobi | 2021-02-12 | clean repo and adapt files |
| html | ee1595d | toobiwankenobi | 2021-02-12 | clean repo and adapt files |
| Rmd | 2e443a5 | toobiwankenobi | 2021-02-09 | remove files that are not needed |
knitr::opts_chunk$set(echo = TRUE, message= FALSE)
knitr::opts_knit$set(root.dir = rprojroot::find_rstudio_root_file())Load libraries
sapply(list.files("code/helper_functions/", full.names = TRUE), source) code/helper_functions//calculateSummary.R
value ?
visible FALSE
code/helper_functions//censor_dat.R
value ?
visible FALSE
code/helper_functions//detect_mRNA_expression.R
value ?
visible FALSE
code/helper_functions//DistanceToClusterCenter.R
value ?
visible FALSE
code/helper_functions//findMilieu.R code/helper_functions//findPatch.R
value ? ?
visible FALSE FALSE
code/helper_functions//getInfoFromString.R
value ?
visible FALSE
code/helper_functions//getSpotnumber.R
value ?
visible FALSE
code/helper_functions//plotCellCounts.R
value ?
visible FALSE
code/helper_functions//plotCellFractions.R
value ?
visible FALSE
code/helper_functions//plotDist.R code/helper_functions//read_Data.R
value ? ?
visible FALSE FALSE
code/helper_functions//scatter_function.R
value ?
visible FALSE
code/helper_functions//sceChecks.R
value ?
visible FALSE
code/helper_functions//validityChecks.R
value ?
visible FALSE library(LSD)
library(SingleCellExperiment)
library(ggplot2)
library(scater)
library(viridis)
library(igraph)
library(CATALYST)
library(reshape2)
library(cowplot)
library(ggridges)
library(pheatmap)
library(tidyverse)Load Data
Cytotoxic T cell Scoring
T Cell Density
We want to calculate the T cell density. we calculate this per mm2. We use the count for cytotxic T cells as defined under “03_cell_type_definition” script
cur_df <- data.frame(celltype = sce_prot$celltype,
ImageNumber = sce_prot$ImageNumber)
# count cell types per images
cellcount <- (t(table(cur_df)))
# here we get the imagesize from the image metadata
im_size <- (image_mat$Height_cellmask * image_mat$Width_cellmask)/1000000
# data frame
cellcount <- data.frame(cellcount)
# we calculate the density for each celltype for 1 mm2
cellcount$density <- cellcount$Freq/im_size[cellcount$ImageNumber]
cellcount <- cellcount[cellcount$celltype == "CD8+ T cell",]
# there are roughly 60 images with 50 or less cytotoxic T cells
hist(cellcount$density,breaks = 300)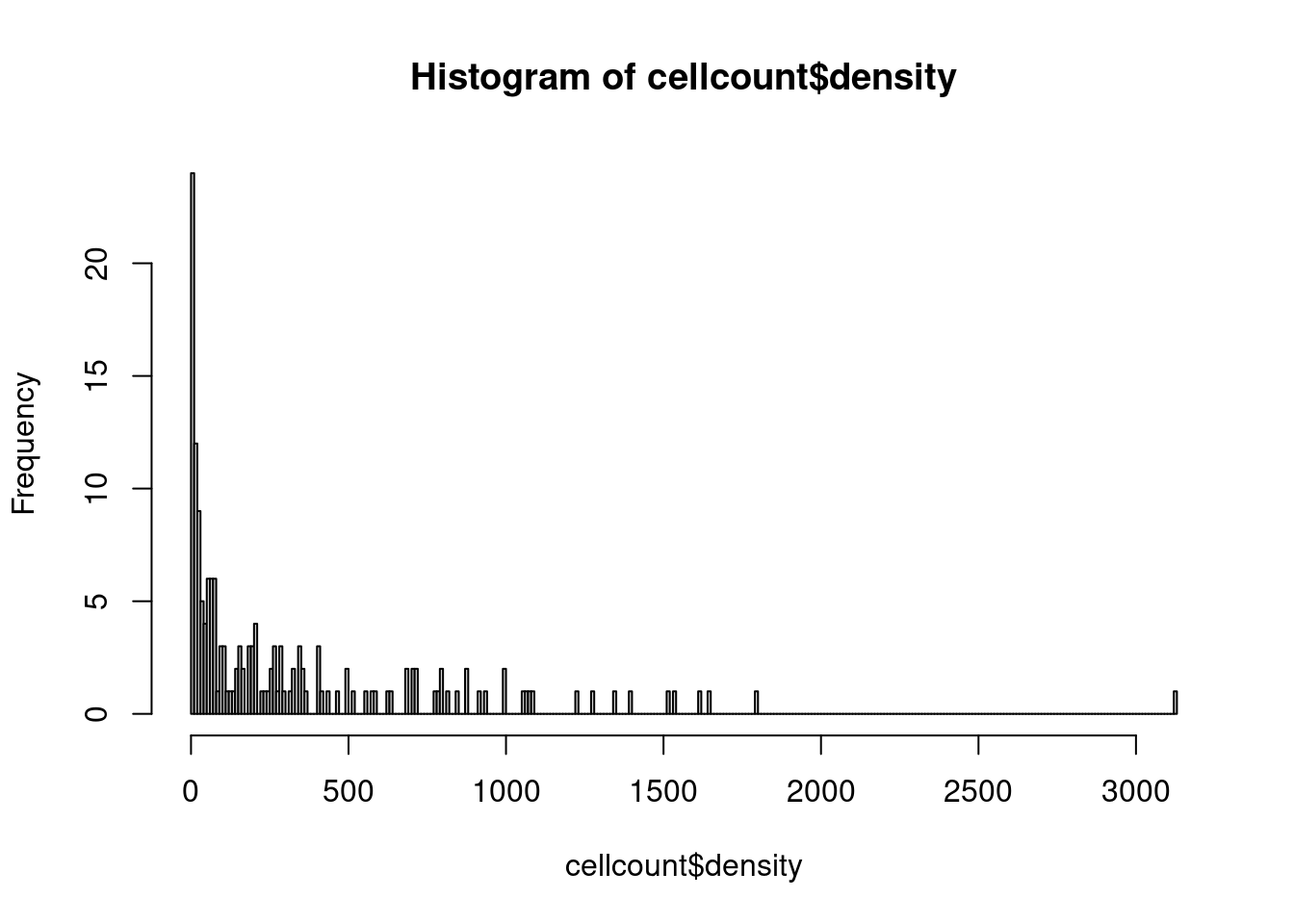
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
# add cyotoxic T cell density to sce
cellcount$ImageNumber <- as.integer(cellcount$ImageNumber)
cur_sce <- data.frame(colData(sce_prot))
cur_sce <- left_join(cur_sce, cellcount[,c("ImageNumber", "density")])
sce_prot$cyotoxic_density_image <- cur_sce$densityT cell density score per image
- absent: 1. quantile
- low: 2. quantile
- med: 3. quantile
- high: 4. quantile
# print quantiles
quantile(cellcount$density) 0% 25% 50% 75% 100%
0.00000 27.07253 145.20423 425.25484 3127.62105 # define a vector with all ImgeNumbers
T_density_scores <- c(1:length(unique(cellcount$ImageNumber)))
T_density_scores <- rep("unassigned",length(unique(cellcount$ImageNumber)))
# use quantiles for scoring system
T_absent <- which(cellcount$density <= quantile(cellcount$density)[[2]])
T_low <- which(cellcount$density > quantile(cellcount$density)[[2]] & cellcount$density <= quantile(cellcount$density)[[3]])
T_med <- which(cellcount$density > quantile(cellcount$density)[[3]] & cellcount$density <= quantile(cellcount$density)[[4]])
T_high <- which(cellcount$density > quantile(cellcount$density)[[4]])
T_density_scores[T_absent] <- "absent"
T_density_scores[T_low] <- "low"
T_density_scores[T_med] <- "med"
T_density_scores[T_high] <- "high"
# now we add the information to the single cell experiment
sce_prot$Tcell_density_score_image <- T_density_scores[sce_prot$ImageNumber]
sce_prot$Tcell_density_score_image <- factor(sce_prot$Tcell_density_score_image, levels = c("absent", "low", "med", "high"))
# number of samples per group
data.frame(colData(sce_prot)) %>%
distinct(Description, .keep_all = T) %>%
group_by(Tcell_density_score_image) %>%
summarise(n=n())# A tibble: 4 × 2
Tcell_density_score_image n
<fct> <int>
1 absent 42
2 low 41
3 med 41
4 high 42E_I_D score compared to T_frac_score_per_ImageNumber
cur_df <- data.frame(T_density = sce_prot$Tcell_density_score_image,
E_I_D = sce_prot$E_I_D)
table(cur_df) E_I_D
T_density D E E/D I I/E
absent 36924 108197 18583 29679 23922
low 8541 64575 4971 65358 53144
med 0 83260 0 84834 63683
high 0 50697 0 133620 131362Add Scores to RNA data set
sce_rna$infiltration <- NULL
sce_rna$T_frac_score_per_BlockID <- NULL
sce_rna$T_frac_score_per_ImageNumber <- NULL
sce_rna$T_frac_score_per_PatientID <- NULL
sce_rna$cyotoxic_density_image <- NULL
description_data <- data.frame(colData(sce_prot)) %>%
distinct(Description, .keep_all = TRUE)
col_rna <- data.frame(colData(sce_rna))
# left_join
col_rna <- left_join(col_rna, description_data[,c("Description", "Tcell_density_score_image", "cyotoxic_density_image")])
# add to sce (attention: cytotoxic density is calculated on protein data set!)
sce_rna$Tcell_density_score_image <- col_rna$Tcell_density_score_image
sce_rna$cyotoxic_density_image <- col_rna$densitySave updated SCE
saveRDS(sce_prot,file = "data/data_for_analysis/sce_protein.rds")
saveRDS(sce_rna,file = "data/data_for_analysis/sce_RNA.rds")
sessionInfo()R version 4.1.2 (2021-11-01)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.3 LTS
Matrix products: default
BLAS/LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.8.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] forcats_0.5.1 stringr_1.4.0
[3] purrr_0.3.4 readr_2.1.2
[5] tidyr_1.2.0 tibble_3.1.6
[7] tidyverse_1.3.1 pheatmap_1.0.12
[9] ggridges_0.5.3 cowplot_1.1.1
[11] reshape2_1.4.4 CATALYST_1.18.1
[13] igraph_1.2.11 viridis_0.6.2
[15] viridisLite_0.4.0 scater_1.22.0
[17] scuttle_1.4.0 ggplot2_3.3.5
[19] SingleCellExperiment_1.16.0 SummarizedExperiment_1.24.0
[21] Biobase_2.54.0 GenomicRanges_1.46.1
[23] GenomeInfoDb_1.30.1 IRanges_2.28.0
[25] S4Vectors_0.32.3 BiocGenerics_0.40.0
[27] MatrixGenerics_1.6.0 matrixStats_0.61.0
[29] LSD_4.1-0 dplyr_1.0.7
[31] workflowr_1.7.0
loaded via a namespace (and not attached):
[1] utf8_1.2.2 tidyselect_1.1.1
[3] grid_4.1.2 BiocParallel_1.28.3
[5] Rtsne_0.15 aws.signature_0.6.0
[7] flowCore_2.6.0 munsell_0.5.0
[9] ScaledMatrix_1.2.0 codetools_0.2-18
[11] withr_2.4.3 colorspace_2.0-2
[13] highr_0.9 knitr_1.37
[15] rstudioapi_0.13 ggsignif_0.6.3
[17] git2r_0.29.0 GenomeInfoDbData_1.2.7
[19] polyclip_1.10-0 farver_2.1.0
[21] flowWorkspace_4.6.0 rprojroot_2.0.2
[23] vctrs_0.3.8 generics_0.1.2
[25] TH.data_1.1-0 xfun_0.29
[27] R6_2.5.1 doParallel_1.0.16
[29] ggbeeswarm_0.6.0 clue_0.3-60
[31] rsvd_1.0.5 bitops_1.0-7
[33] DelayedArray_0.20.0 assertthat_0.2.1
[35] promises_1.2.0.1 scales_1.1.1
[37] multcomp_1.4-18 beeswarm_0.4.0
[39] gtable_0.3.0 beachmat_2.10.0
[41] processx_3.5.2 RProtoBufLib_2.6.0
[43] sandwich_3.0-1 rlang_1.0.0
[45] GlobalOptions_0.1.2 splines_4.1.2
[47] rstatix_0.7.0 hexbin_1.28.2
[49] broom_0.7.12 modelr_0.1.8
[51] yaml_2.2.2 abind_1.4-5
[53] backports_1.4.1 httpuv_1.6.5
[55] RBGL_1.70.0 tools_4.1.2
[57] ellipsis_0.3.2 jquerylib_0.1.4
[59] RColorBrewer_1.1-2 Rcpp_1.0.8
[61] plyr_1.8.6 base64enc_0.1-3
[63] sparseMatrixStats_1.6.0 zlibbioc_1.40.0
[65] RCurl_1.98-1.5 ps_1.6.0
[67] FlowSOM_2.2.0 ggpubr_0.4.0
[69] GetoptLong_1.0.5 zoo_1.8-9
[71] haven_2.4.3 ggrepel_0.9.1
[73] cluster_2.1.2 colorRamps_2.3
[75] fs_1.5.2 magrittr_2.0.2
[77] ncdfFlow_2.40.0 data.table_1.14.2
[79] scattermore_0.7 circlize_0.4.13
[81] reprex_2.0.1 mvtnorm_1.1-3
[83] whisker_0.4 ggnewscale_0.4.5
[85] hms_1.1.1 evaluate_0.14
[87] XML_3.99-0.8 jpeg_0.1-9
[89] readxl_1.3.1 gridExtra_2.3
[91] shape_1.4.6 ggcyto_1.22.0
[93] compiler_4.1.2 crayon_1.4.2
[95] ggpointdensity_0.1.0 htmltools_0.5.2
[97] tzdb_0.2.0 later_1.3.0
[99] RcppParallel_5.1.5 lubridate_1.8.0
[101] aws.s3_0.3.21 DBI_1.1.2
[103] tweenr_1.0.2 dbplyr_2.1.1
[105] ComplexHeatmap_2.10.0 MASS_7.3-55
[107] Matrix_1.4-0 car_3.0-12
[109] cli_3.1.1 parallel_4.1.2
[111] pkgconfig_2.0.3 getPass_0.2-2
[113] xml2_1.3.3 foreach_1.5.2
[115] vipor_0.4.5 bslib_0.3.1
[117] XVector_0.34.0 drc_3.0-1
[119] rvest_1.0.2 callr_3.7.0
[121] digest_0.6.29 ConsensusClusterPlus_1.58.0
[123] graph_1.72.0 cellranger_1.1.0
[125] rmarkdown_2.11 DelayedMatrixStats_1.16.0
[127] curl_4.3.2 gtools_3.9.2
[129] rjson_0.2.21 lifecycle_1.0.1
[131] jsonlite_1.7.3 carData_3.0-5
[133] BiocNeighbors_1.12.0 fansi_1.0.2
[135] pillar_1.7.0 lattice_0.20-45
[137] plotrix_3.8-2 fastmap_1.1.0
[139] httr_1.4.2 survival_3.2-13
[141] glue_1.6.1 png_0.1-7
[143] iterators_1.0.13 Rgraphviz_2.38.0
[145] nnls_1.4 ggforce_0.3.3
[147] stringi_1.7.6 sass_0.4.0
[149] BiocSingular_1.10.0 CytoML_2.6.0
[151] latticeExtra_0.6-29 cytolib_2.6.1
[153] irlba_2.3.5