13_Dysfunction Stain
2021-01-19
Last updated: 2022-02-22
Checks: 7 0
Knit directory: MelanomaIMC/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200728) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version d246c15. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rproj.user/
Ignored: Table_S4.csv
Ignored: code/.DS_Store
Ignored: code/._.DS_Store
Ignored: data/.DS_Store
Ignored: data/._.DS_Store
Ignored: data/data_for_analysis/
Ignored: data/full_data/
Unstaged changes:
Modified: .gitignore
Modified: analysis/Supp-Figure_10.rmd
Modified: analysis/_site.yml
Deleted: analysis/license.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/13_Dysfunction_Stain.rmd) and HTML (docs/13_Dysfunction_Stain.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 73aa800 | toobiwankenobi | 2022-02-22 | add .html for static website |
| Rmd | fa0f601 | toobiwankenobi | 2022-02-06 | clean Supp Fig code |
Introduction
Here, we load the data from the dysfunction stain. Images with a T cell dysfunction score were re-acquired with an extended T cell marker panel. An SCE object will be created and saved at the end and serves for further analyses (e.g. Fig 4D, Fig S10B)
Preparation
Load Libraries
library(SingleCellExperiment)Loading required package: SummarizedExperimentLoading required package: MatrixGenericsLoading required package: matrixStats
Attaching package: 'MatrixGenerics'The following objects are masked from 'package:matrixStats':
colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
colWeightedMeans, colWeightedMedians, colWeightedSds,
colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
rowWeightedSds, rowWeightedVarsLoading required package: GenomicRangesLoading required package: stats4Loading required package: BiocGenerics
Attaching package: 'BiocGenerics'The following objects are masked from 'package:stats':
IQR, mad, sd, var, xtabsThe following objects are masked from 'package:base':
anyDuplicated, append, as.data.frame, basename, cbind, colnames,
dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget,
order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply,
union, unique, unsplit, which.max, which.minLoading required package: S4Vectors
Attaching package: 'S4Vectors'The following objects are masked from 'package:base':
expand.grid, I, unnameLoading required package: IRangesLoading required package: GenomeInfoDbLoading required package: BiobaseWelcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Attaching package: 'Biobase'The following object is masked from 'package:MatrixGenerics':
rowMediansThe following objects are masked from 'package:matrixStats':
anyMissing, rowMedianslibrary(imcRtools)Loading required package: SpatialExperimentlibrary(BiocParallel)
library(BiocNeighbors)
library(dittoSeq)Loading required package: ggplot2library(scater)Loading required package: scuttlelibrary(scales)
library(cowplot)
library(Hmisc)Loading required package: latticeLoading required package: survivalLoading required package: Formula
Attaching package: 'Hmisc'The following object is masked from 'package:Biobase':
contentsThe following objects are masked from 'package:base':
format.pval, unitslibrary(tidyverse)── Attaching packages ─────────────────────────────────────── tidyverse 1.3.1 ──✓ tibble 3.1.6 ✓ dplyr 1.0.7
✓ tidyr 1.2.0 ✓ stringr 1.4.0
✓ readr 2.1.2 ✓ forcats 0.5.1
✓ purrr 0.3.4 ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
x readr::col_factor() masks scales::col_factor()
x dplyr::collapse() masks IRanges::collapse()
x dplyr::combine() masks Biobase::combine(), BiocGenerics::combine()
x dplyr::count() masks matrixStats::count()
x dplyr::desc() masks IRanges::desc()
x purrr::discard() masks scales::discard()
x tidyr::expand() masks S4Vectors::expand()
x dplyr::filter() masks stats::filter()
x dplyr::first() masks S4Vectors::first()
x dplyr::lag() masks stats::lag()
x ggplot2::Position() masks BiocGenerics::Position(), base::Position()
x purrr::reduce() masks GenomicRanges::reduce(), IRanges::reduce()
x dplyr::rename() masks S4Vectors::rename()
x dplyr::slice() masks IRanges::slice()
x dplyr::src() masks Hmisc::src()
x dplyr::summarize() masks Hmisc::summarize()library(ggpubr)
Attaching package: 'ggpubr'The following object is masked from 'package:cowplot':
get_legendlibrary(scran)
set.seed(12345)Load Data
sce_1 <- read_steinbock(path = "data/full_data/exhaustion_stain/",
return_as = "sce", graphs_folder = NULL, regionprops_folder = NULL,
image_file = NULL)
sce_2 <- read_steinbock(path = "data/full_data/revision_stain/",
return_as = "sce", graphs_folder = NULL, regionprops_folder = NULL,
image_file = NULL)
sce_RNA <- readRDS(file = "data/data_for_analysis/sce_RNA.rds")add metadata to sce_1
assay(sce_1,"asinh") <- asinh(counts(sce_1))
colnames(sce_1) <- paste0(sce_1$sample_id,"_",sce_1$ObjectNumber)
# add information from dysfunction score and Description
imnames <- c("E5","H9","F9","L8","N10","F2","D8","E4","J4","G11","L4","O9")
names(imnames) <- unique(sce_1$sample_id)
sce_1$Description <- imnames[sce_1$sample_id]
# Dysfunction Score
highDys <- unique(sce_RNA[,sce_RNA$dysfunction_score %in% "High Dysfunction"]$Description)
lowDys <- unique(sce_RNA[,sce_RNA$dysfunction_score %in% "Low Dysfunction"]$Description)
sce_1$dysfunction_score <- NA
sce_1[,sce_1$Description %in% highDys]$dysfunction_score <- "High Dysfunction"
sce_1[,sce_1$Description %in% lowDys]$dysfunction_score <- "Low Dysfunction"add metadata to sce_2
assay(sce_2,"asinh") <- asinh(counts(sce_2))
colnames(sce_2) <- paste0(sce_2$sample_id,"_",sce_2$ObjectNumber)
# add information from dysfunction score and Description
imnames <- c("E2","D4","D3","G9","B6","O2","M10","B3","N8","A4","I11","K2","I5","M9")
names(imnames) <- unique(sce_2$sample_id)[c(1,7:14,2:6)]
sce_2$Description <- imnames[sce_2$sample_id]
sce_2$dysfunction_score <- NA
sce_2[,sce_2$Description %in% highDys]$dysfunction_score <- "High Dysfunction"
sce_2[,sce_2$Description %in% lowDys]$dysfunction_score <- "Low Dysfunction"merge sce objects
we will also remove images E2. this images was the first image of a measurement after downtime of the machine and had substantially higher intensities that all other images.
sce <- cbind(sce_1,sce_2)
rm(sce_1,sce_2)
sce <- sce[,sce$Description != "E2"]
# remove images with no dysfunction score
sce <- sce[,sce$dysfunction_score %in% c("High Dysfunction", "Low Dysfunction")]Analysis
define cut offs for the definition of CD8 T cells per sample
here we check if there are any global intensity biases for CD8
dittoRidgePlot(sce,var="CD8a", group.by= "Description",assay = "asinh")Picking joint bandwidth of 0.144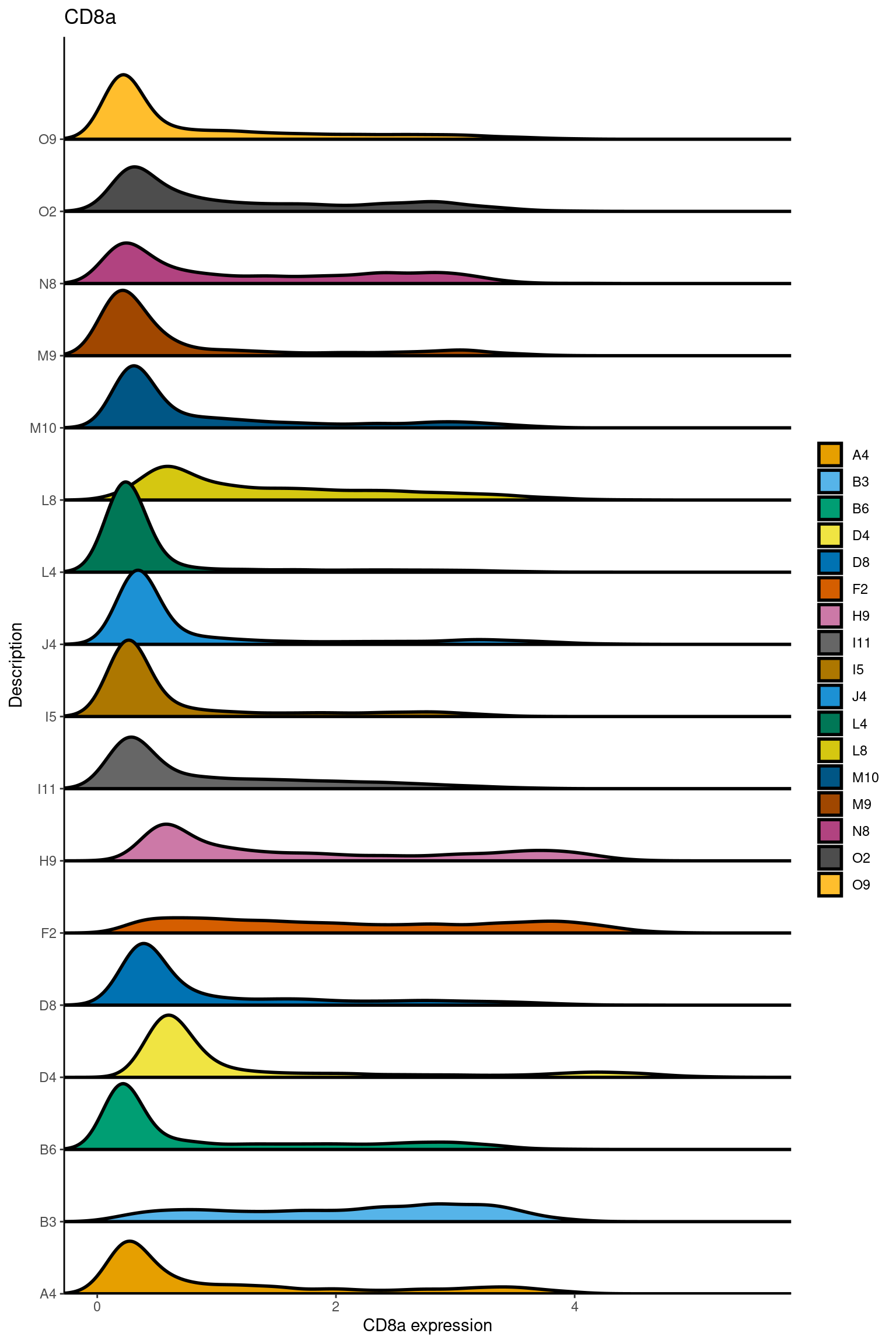
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
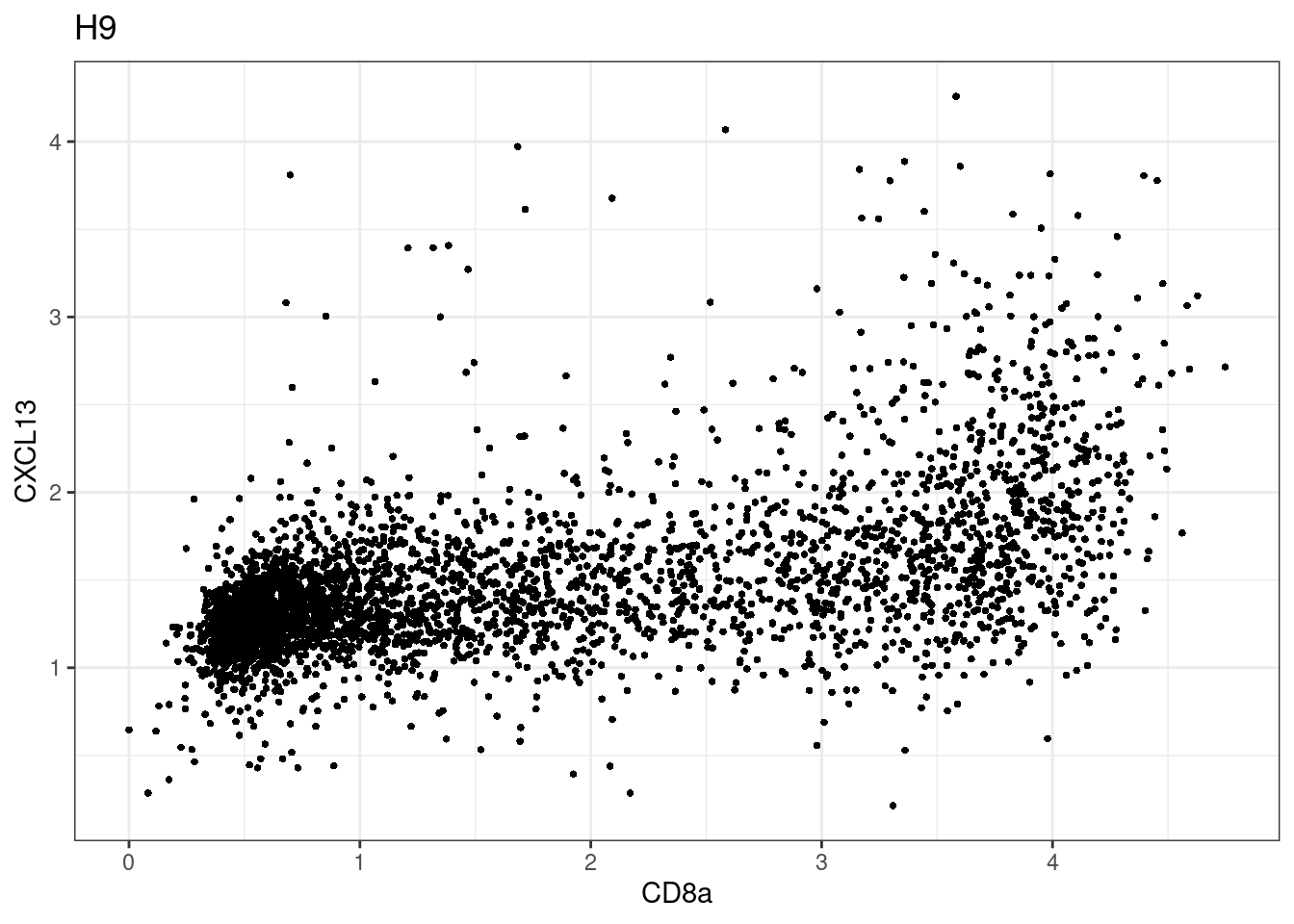
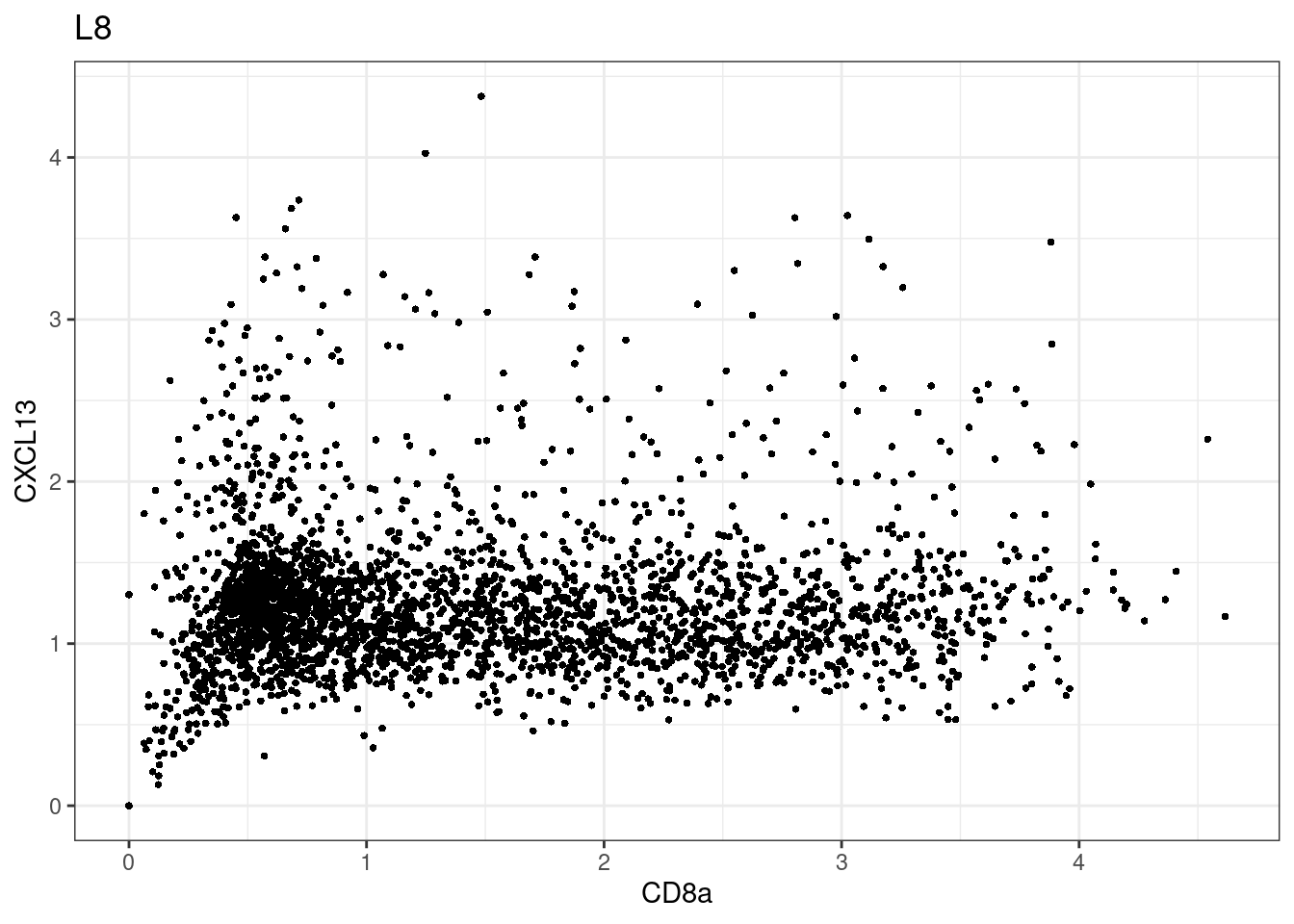
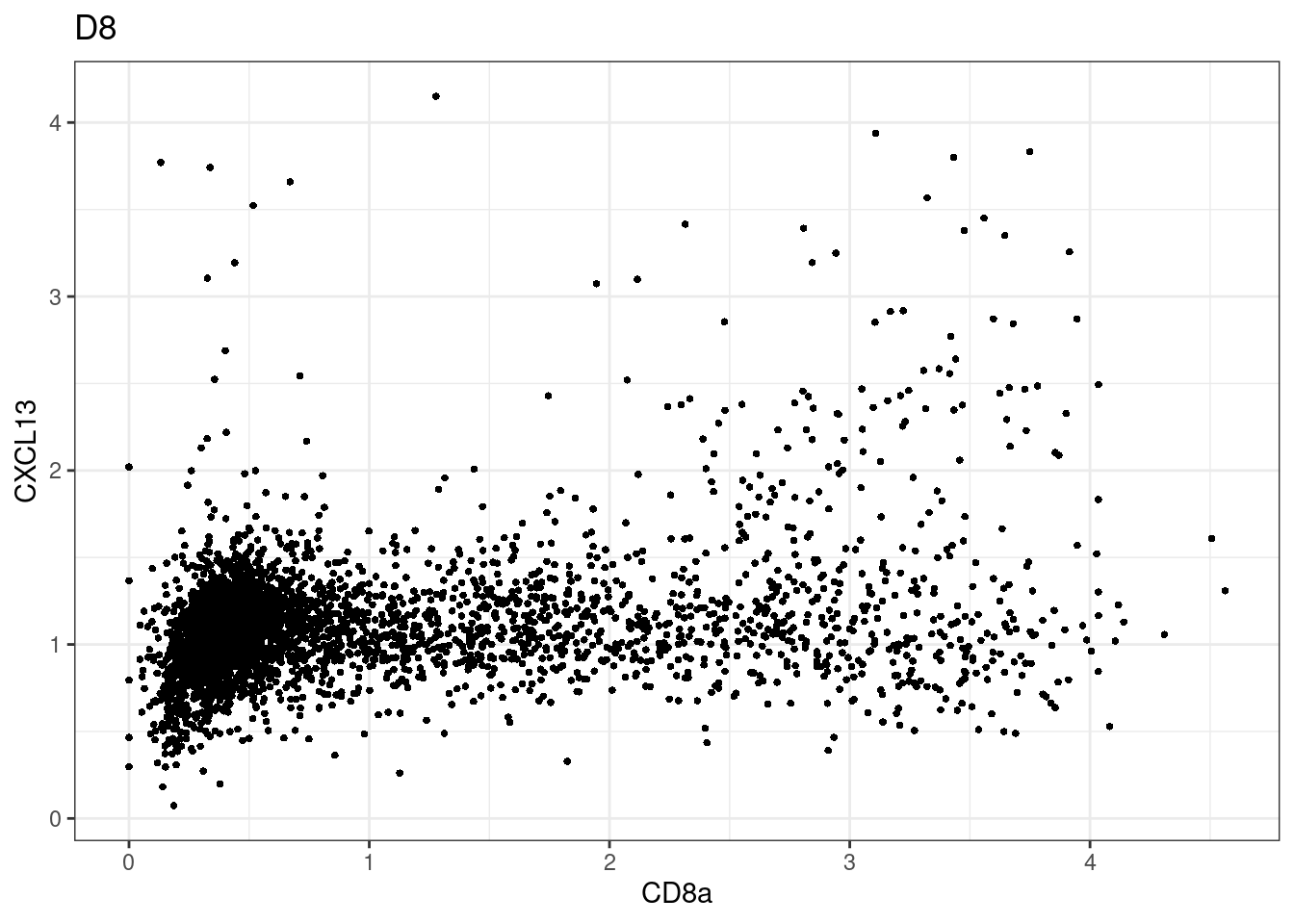
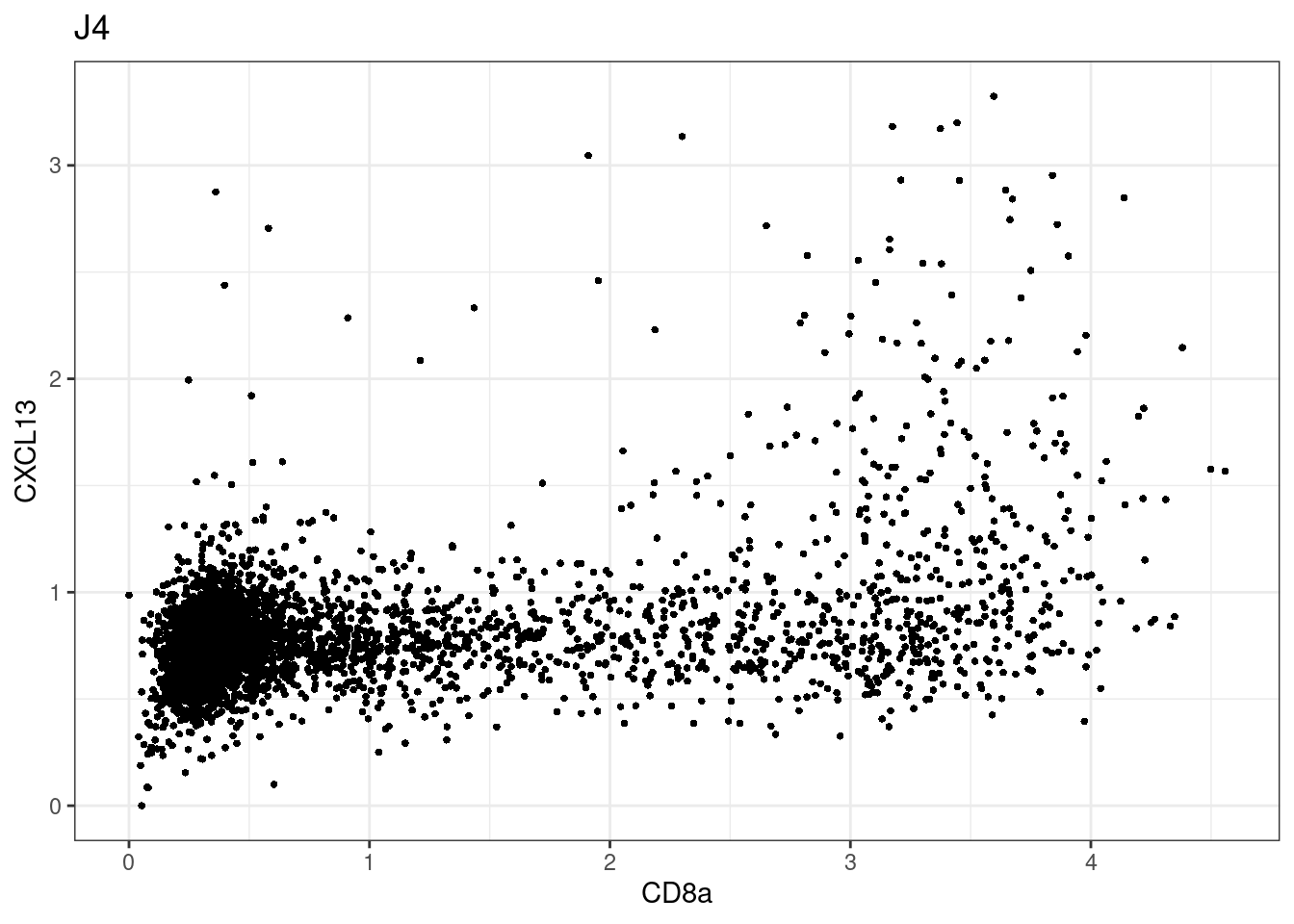
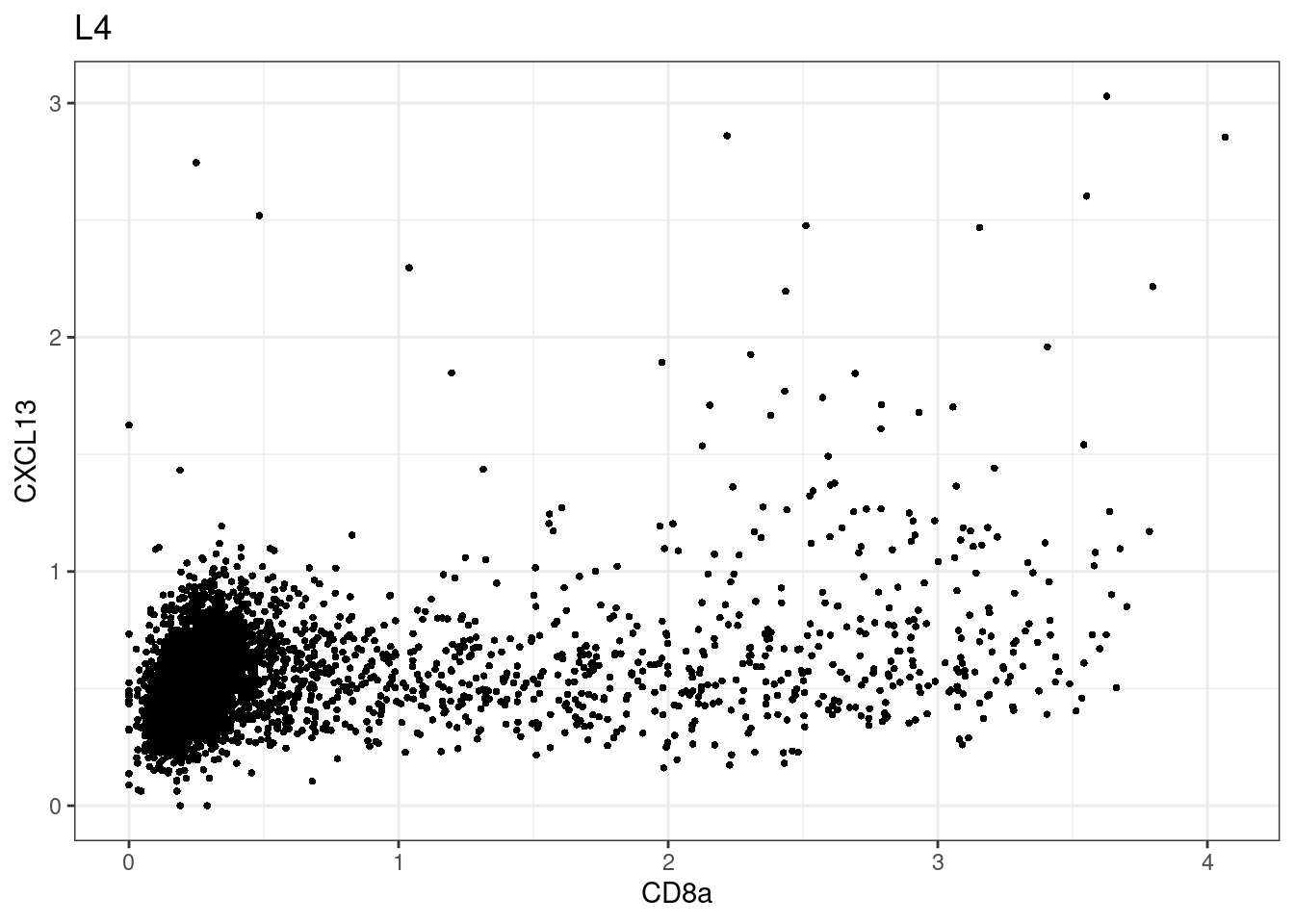
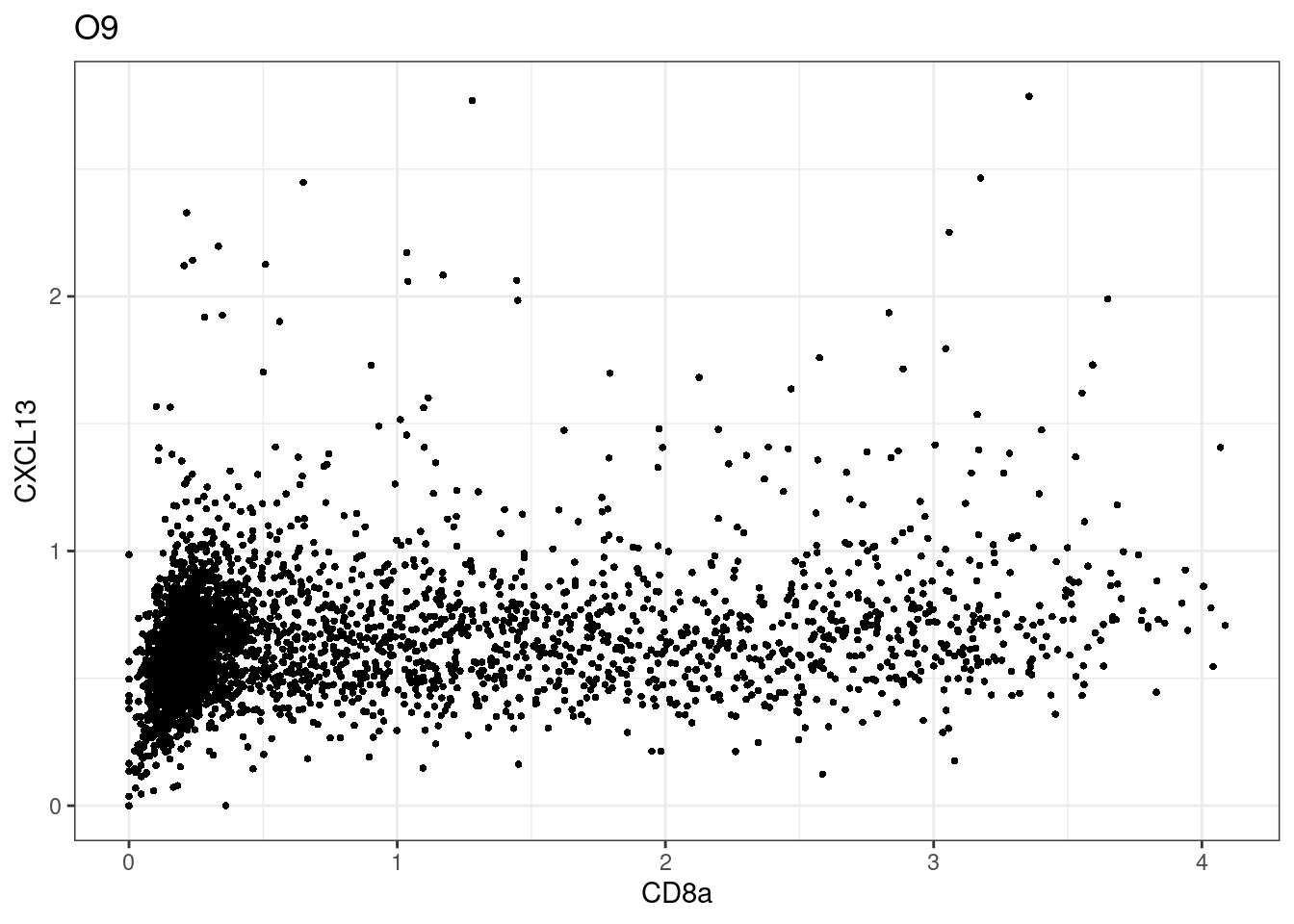
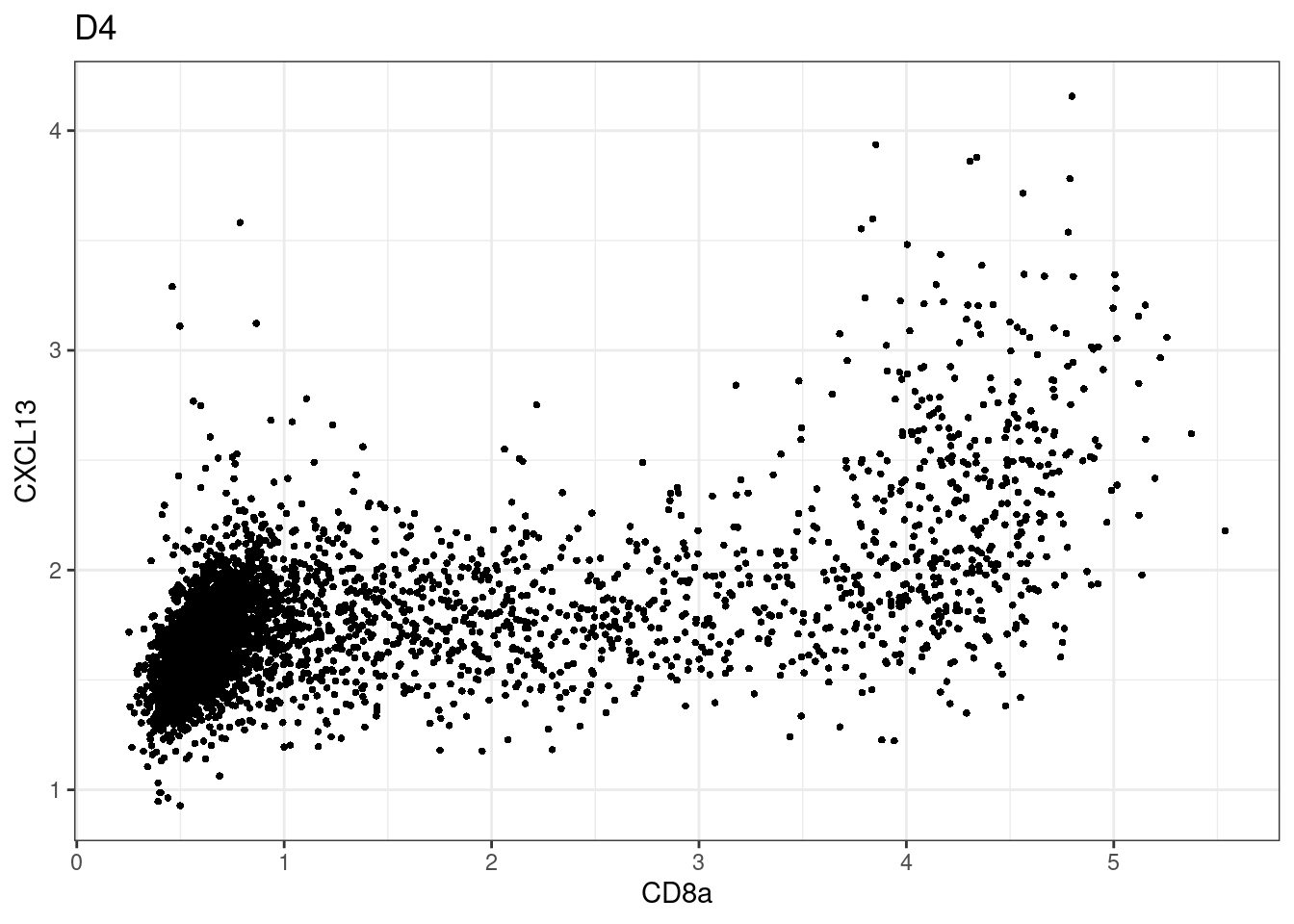
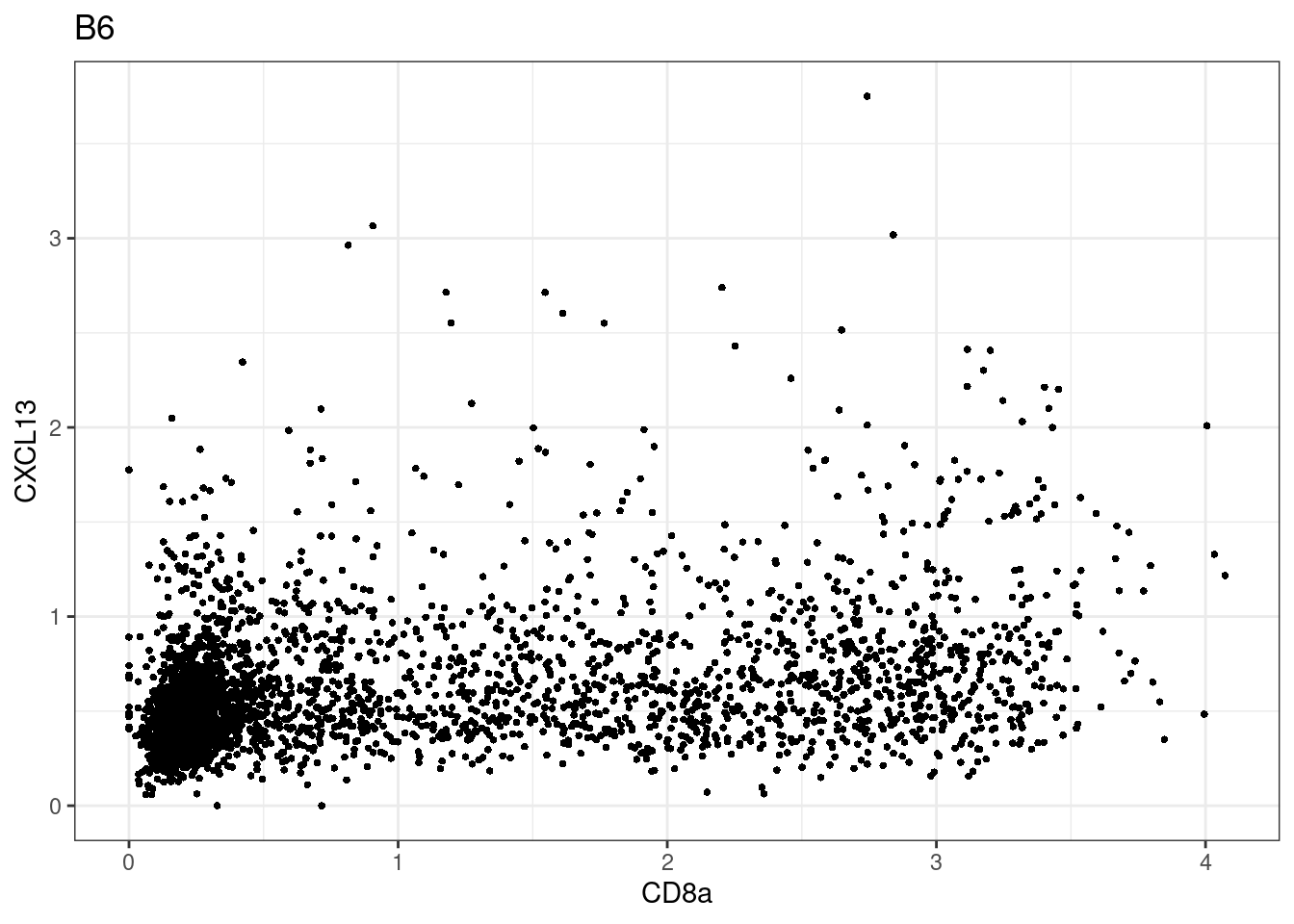
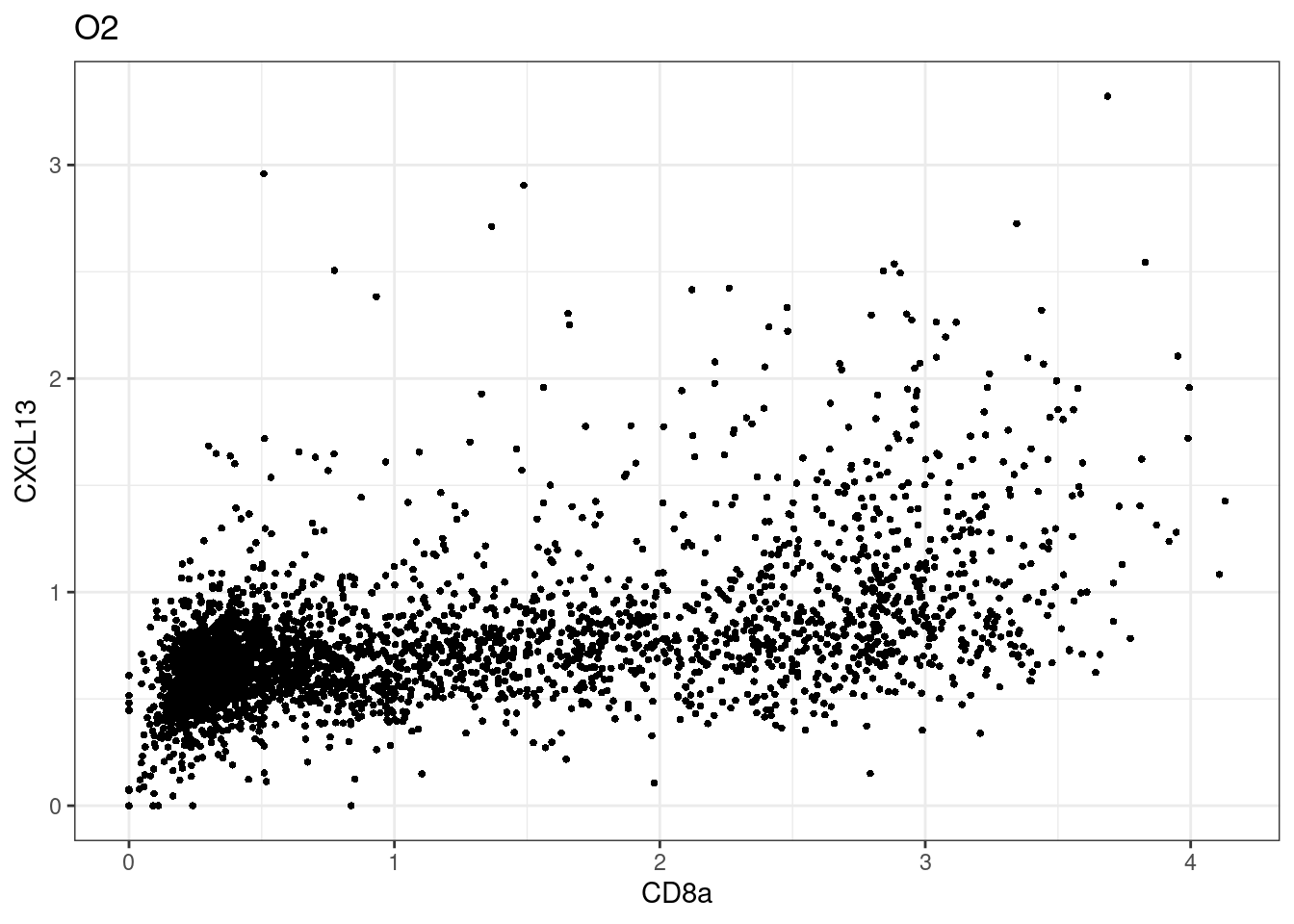
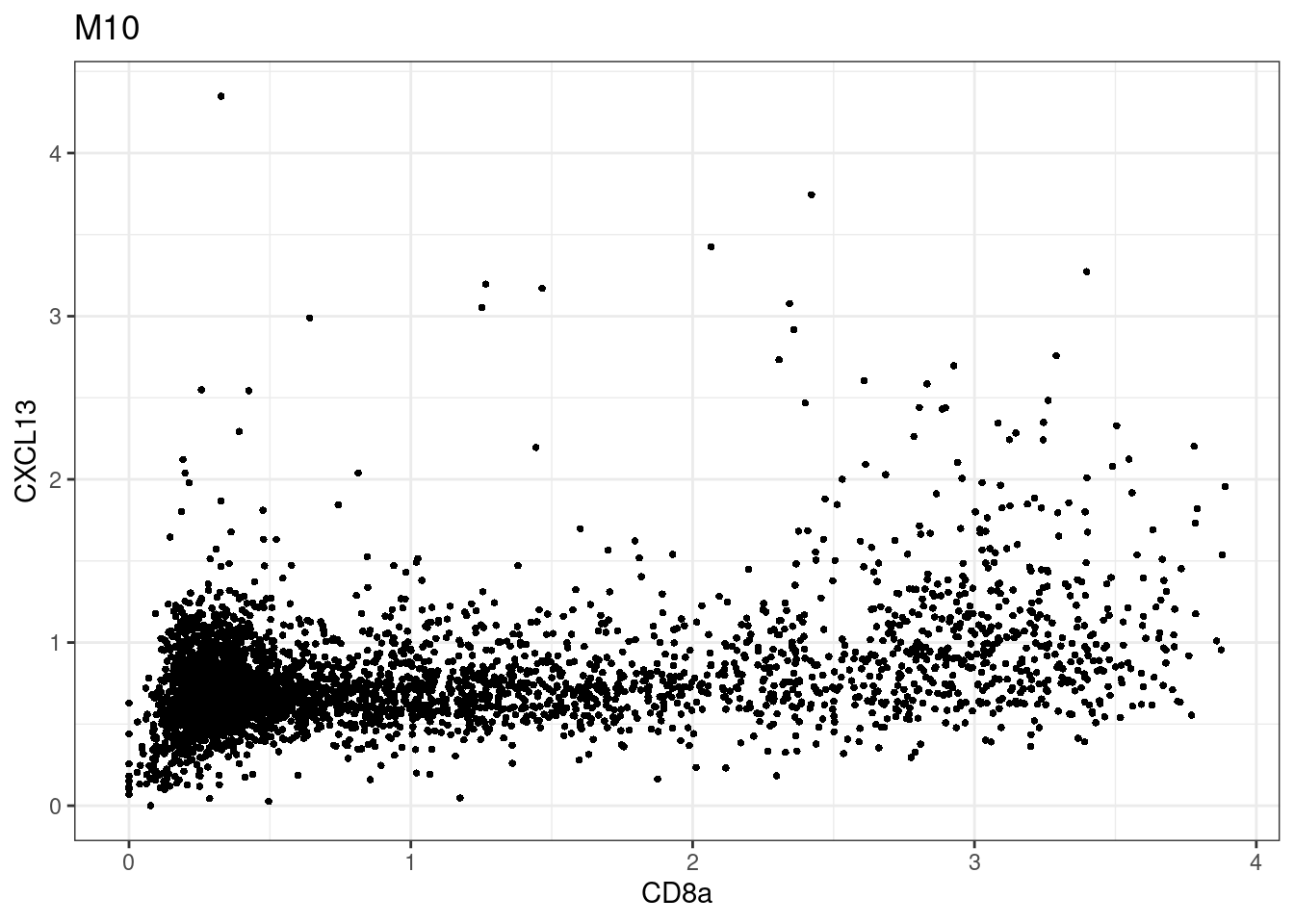
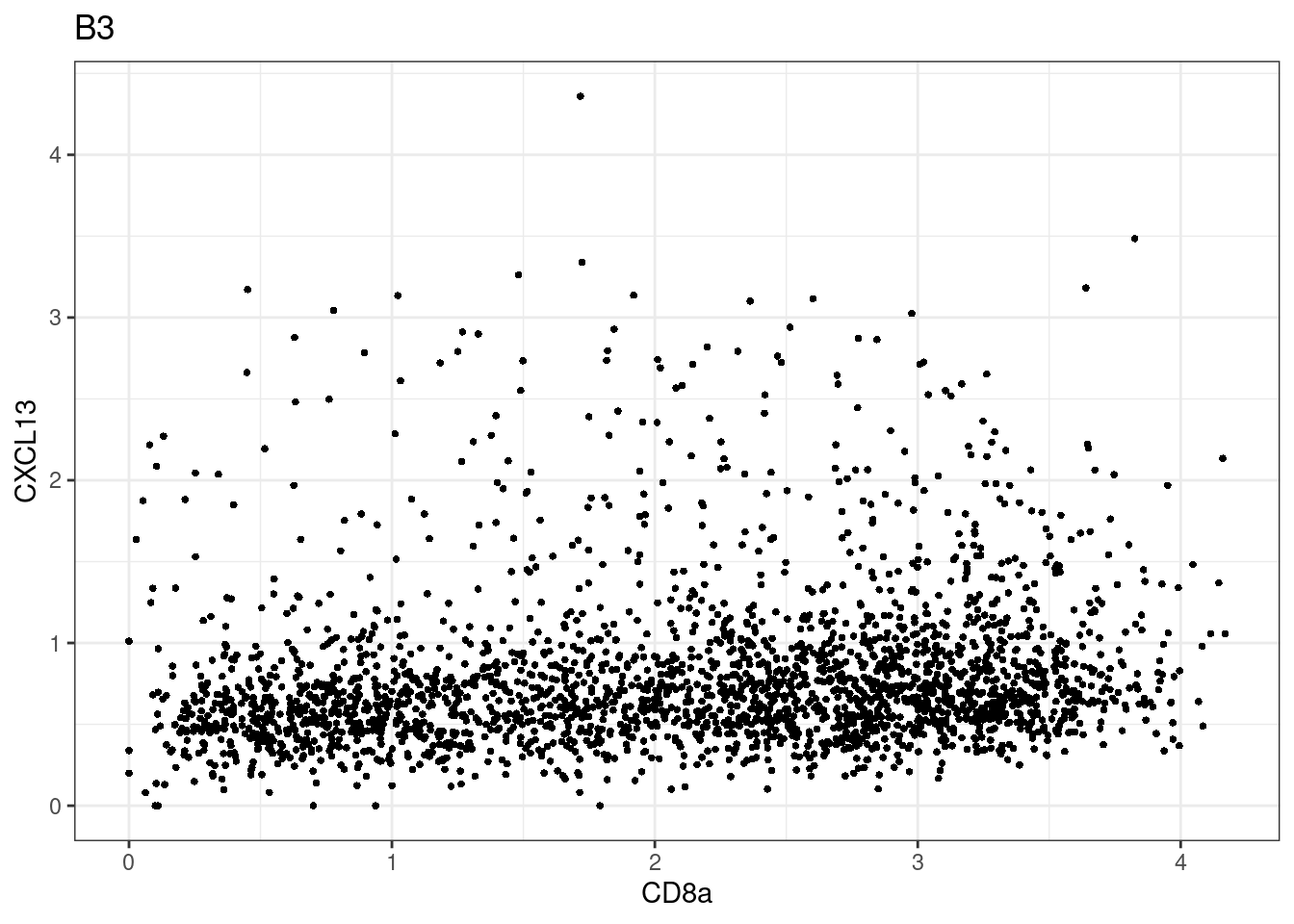
samples E2, F2 L8, D4 and B3 should be inspected further since they have higher CD8 background. However, we will more generally chose a conservative cut off for T cell definition
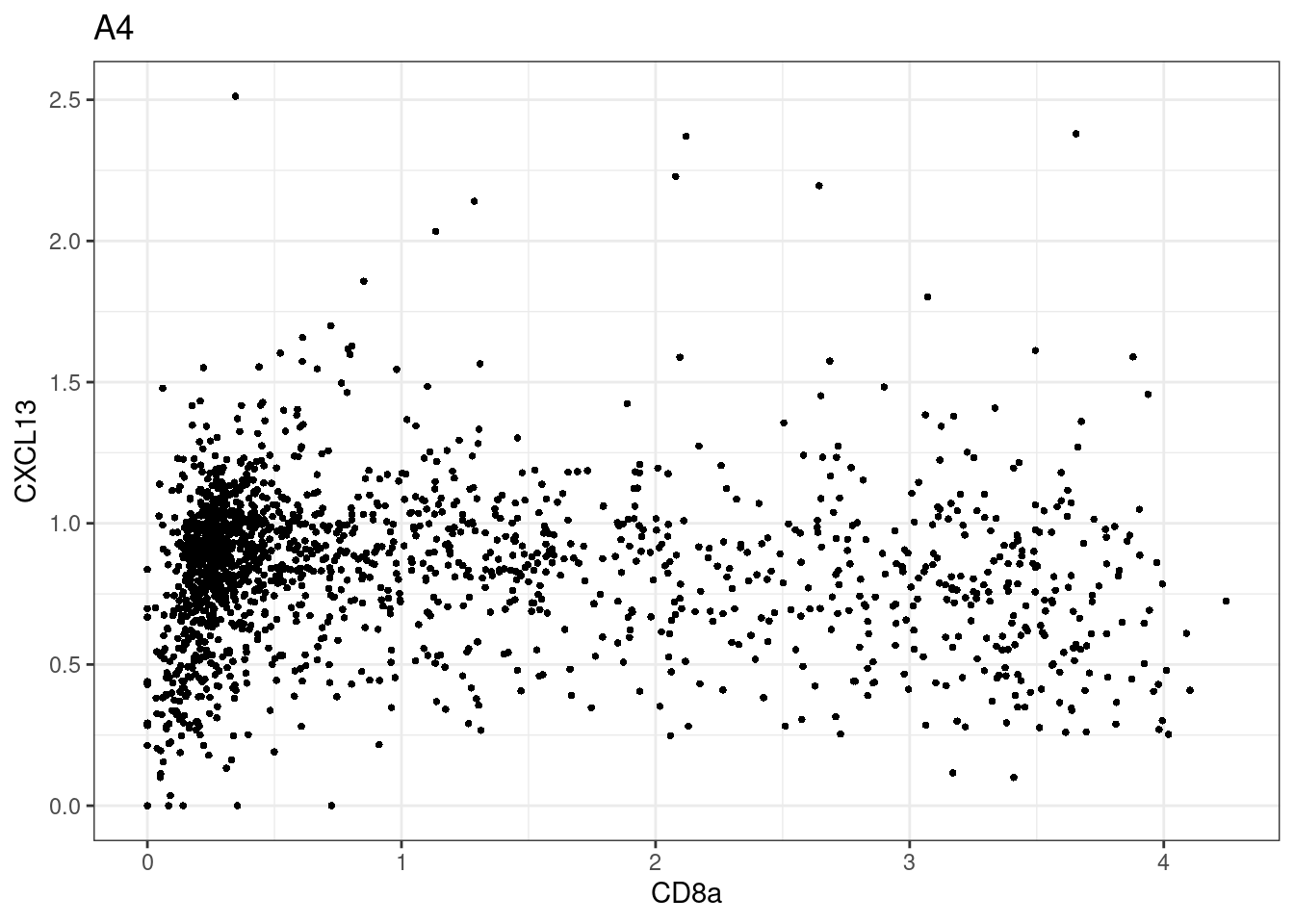
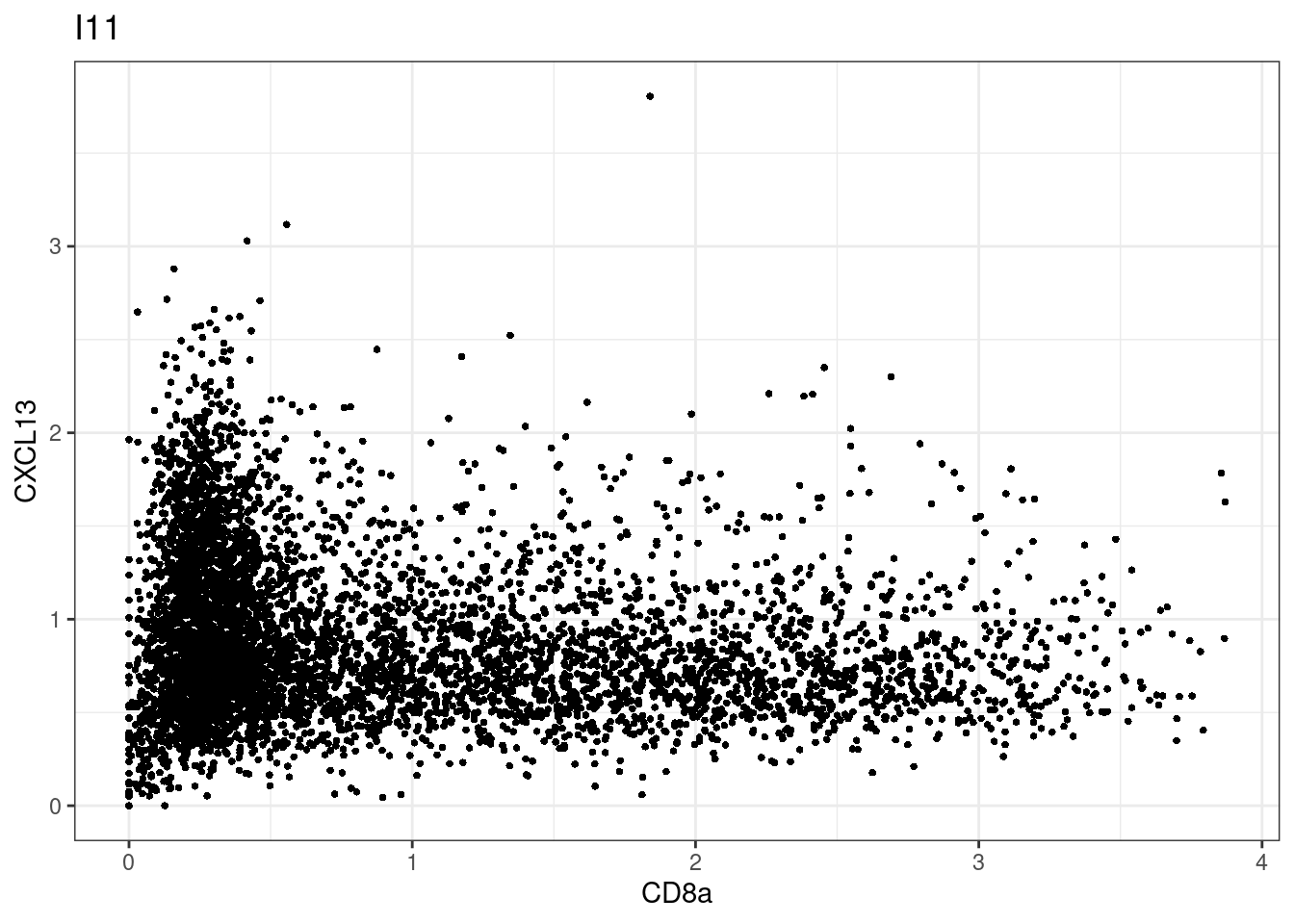
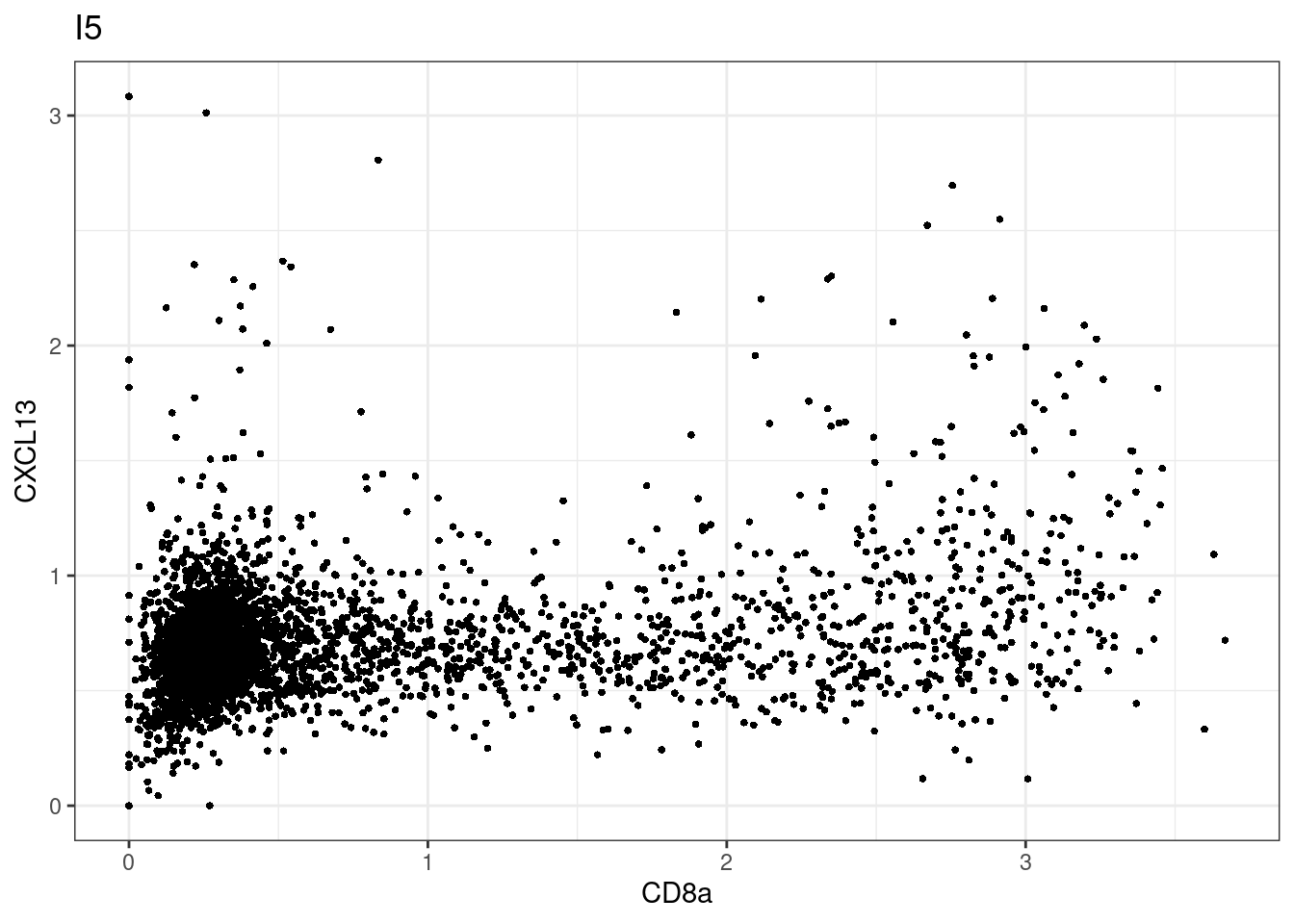
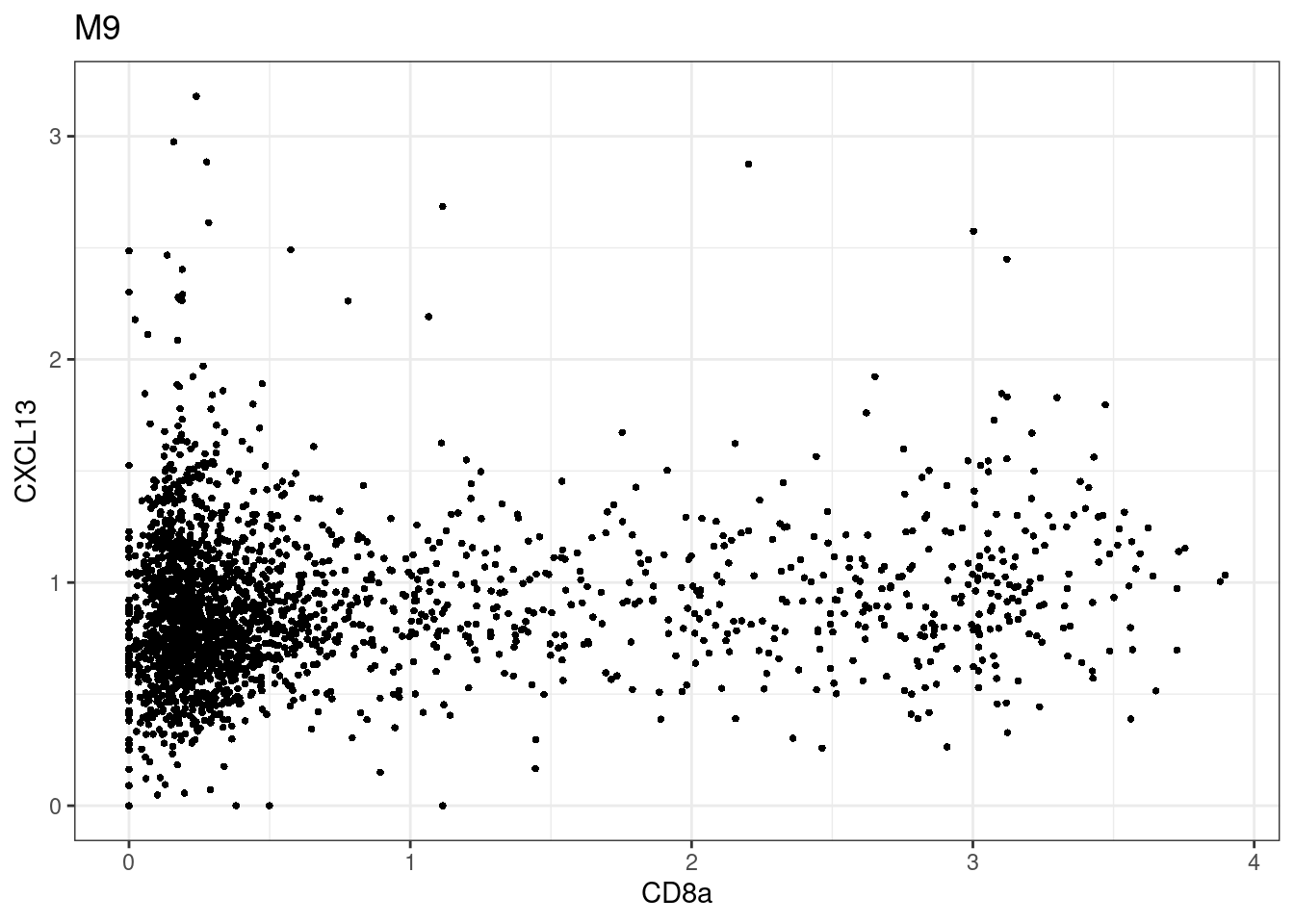
for (i in unique(sce$Description)) {
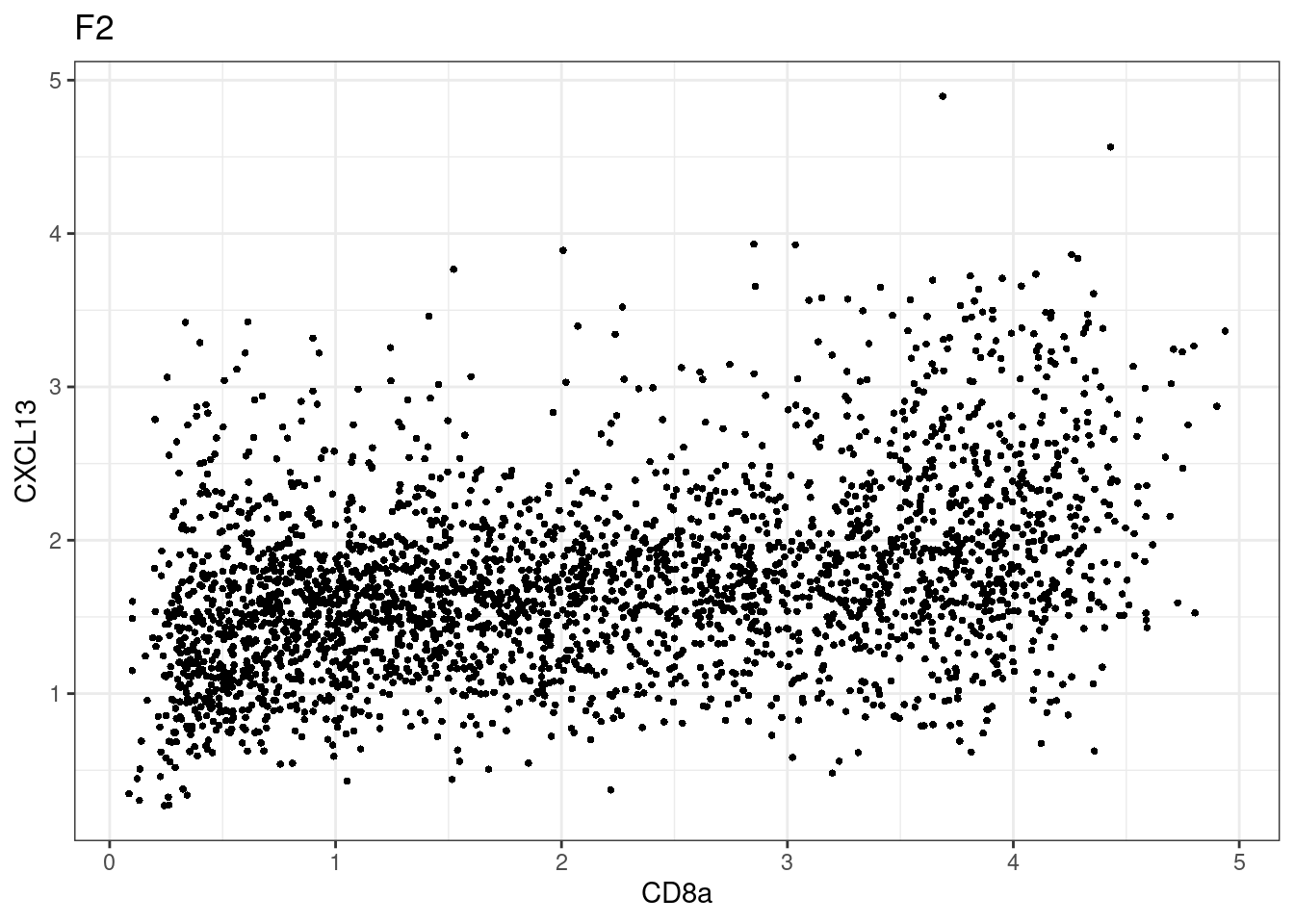
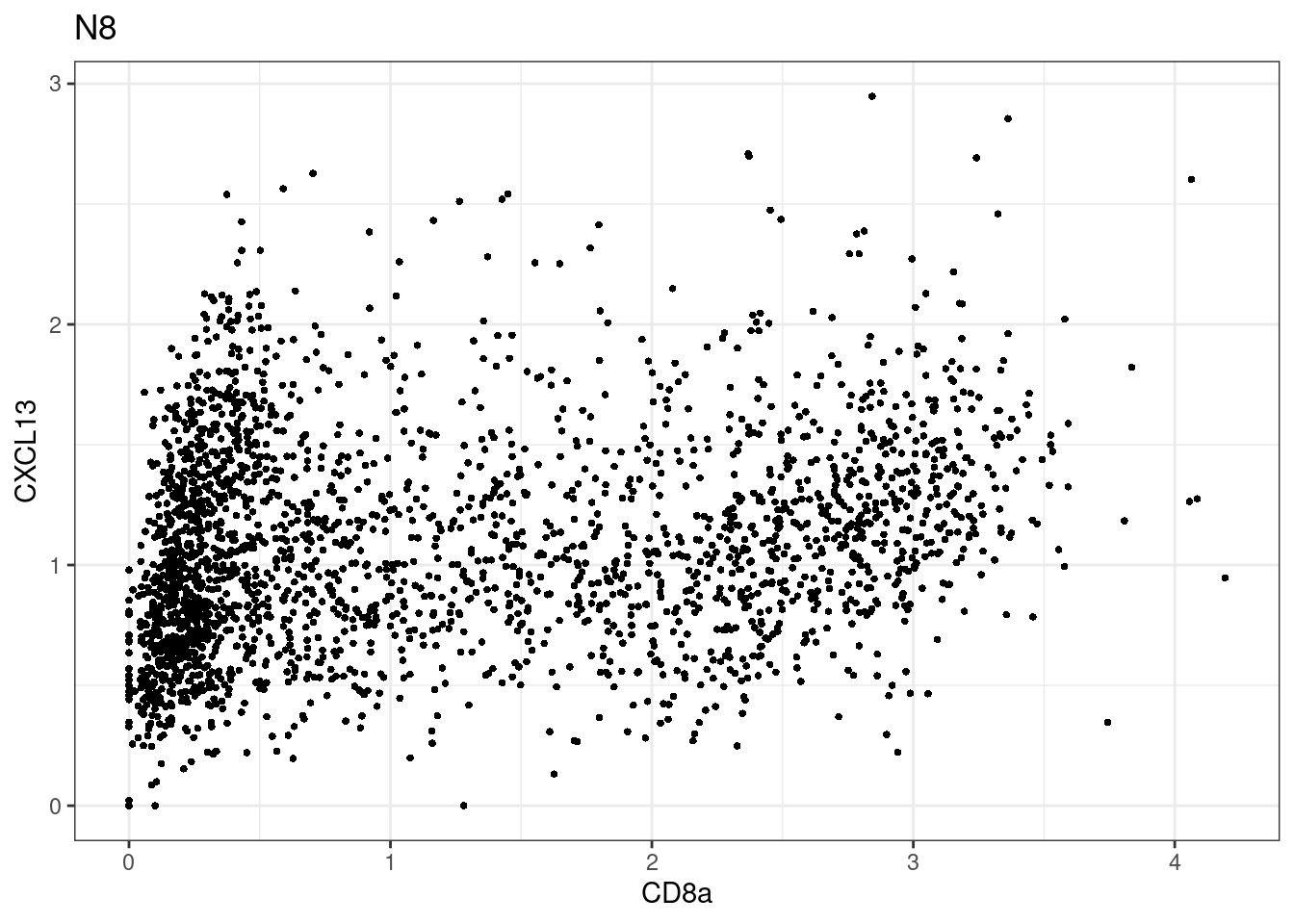
p <-dittoScatterPlot(sce[,which(sce$Description == i )],x.var = "CD8a",y.var = "CXCL13",assay.x = "asinh",assay.y = "asinh", main=i,)
plot(p)
}
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
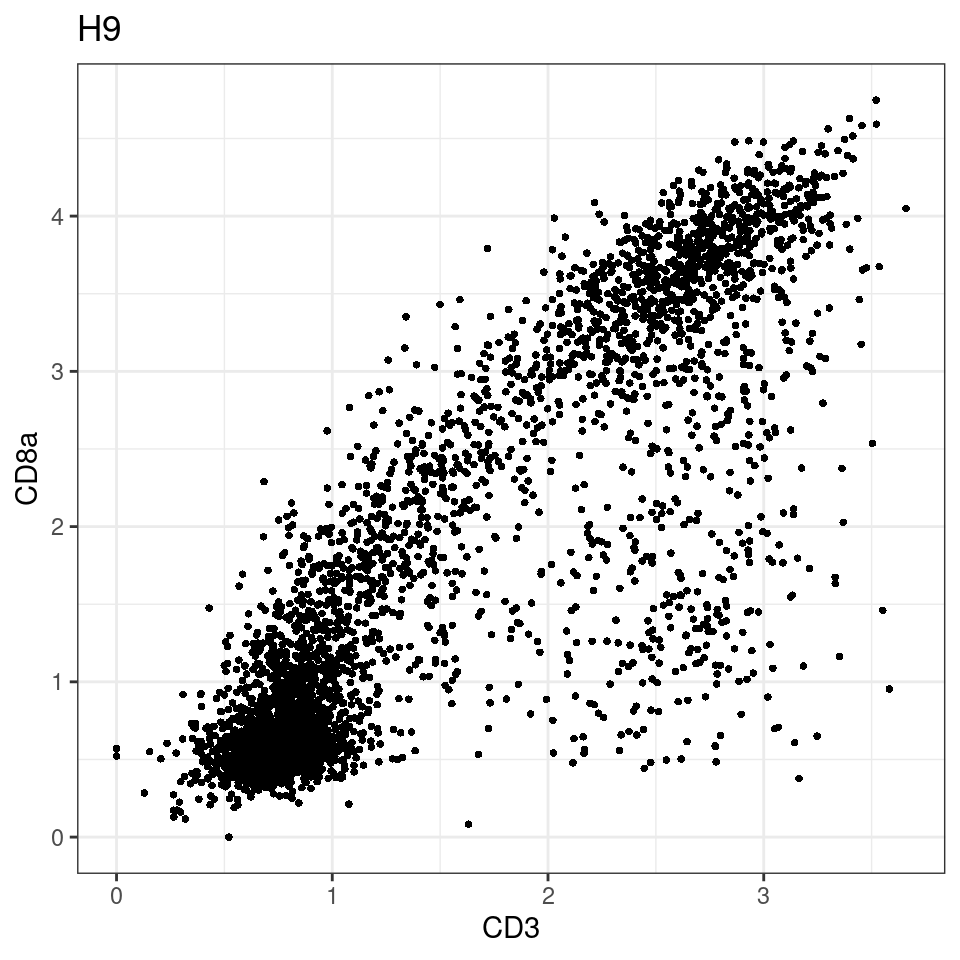
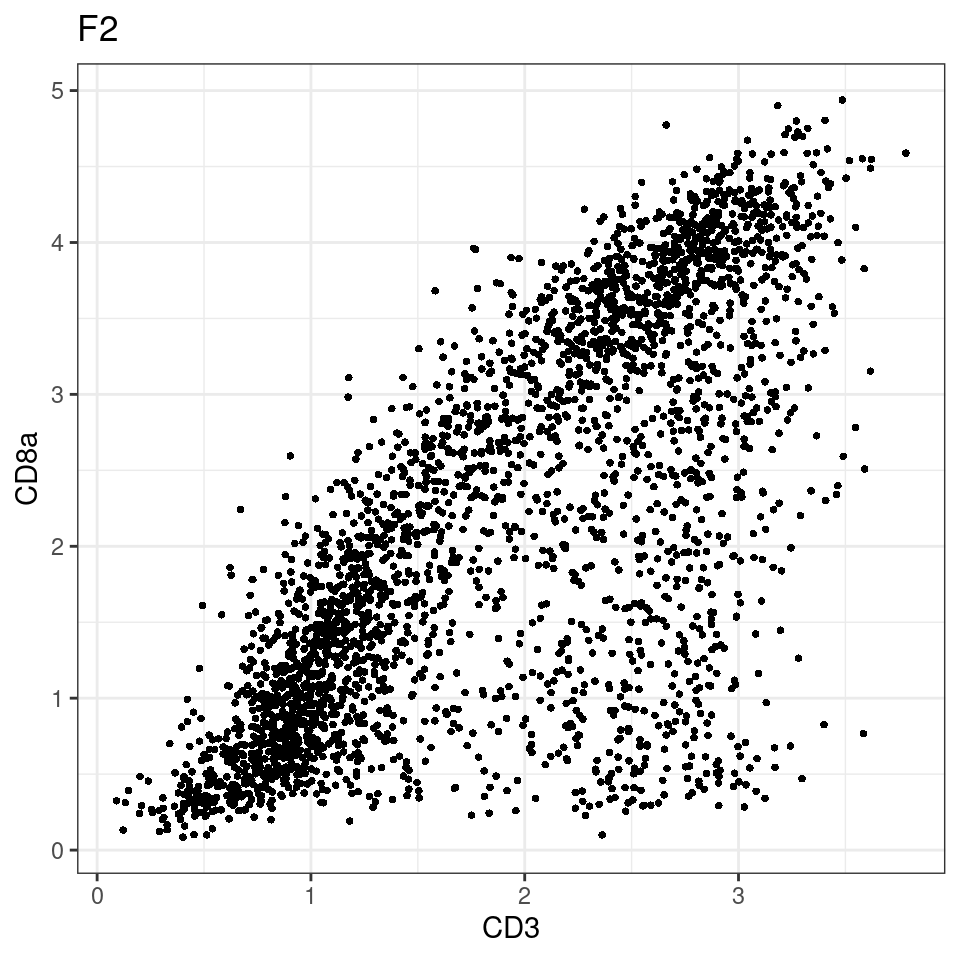
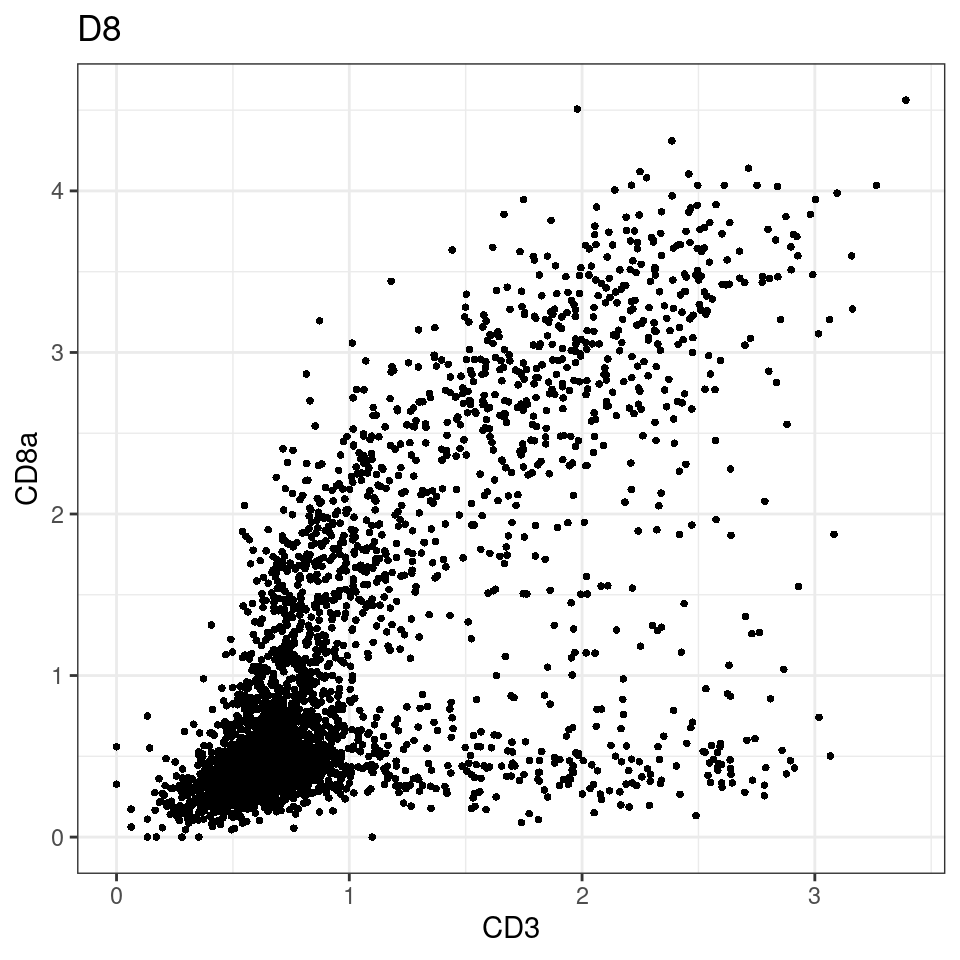
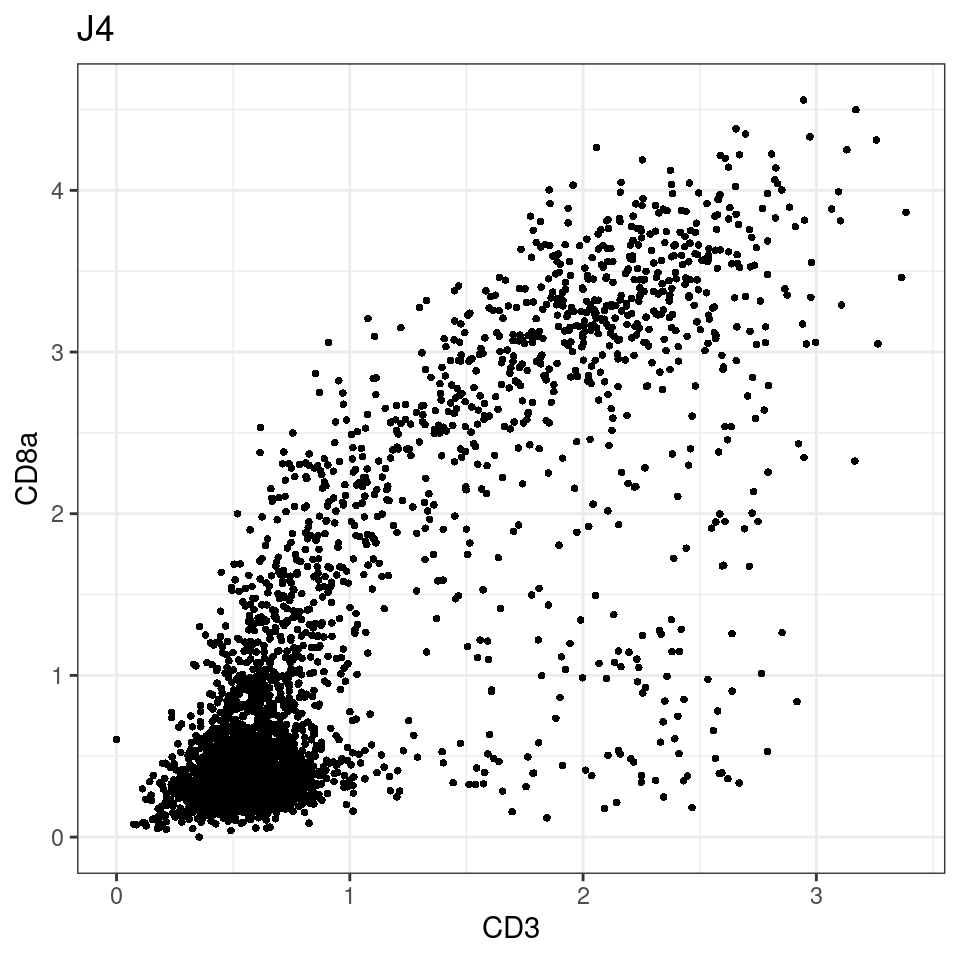
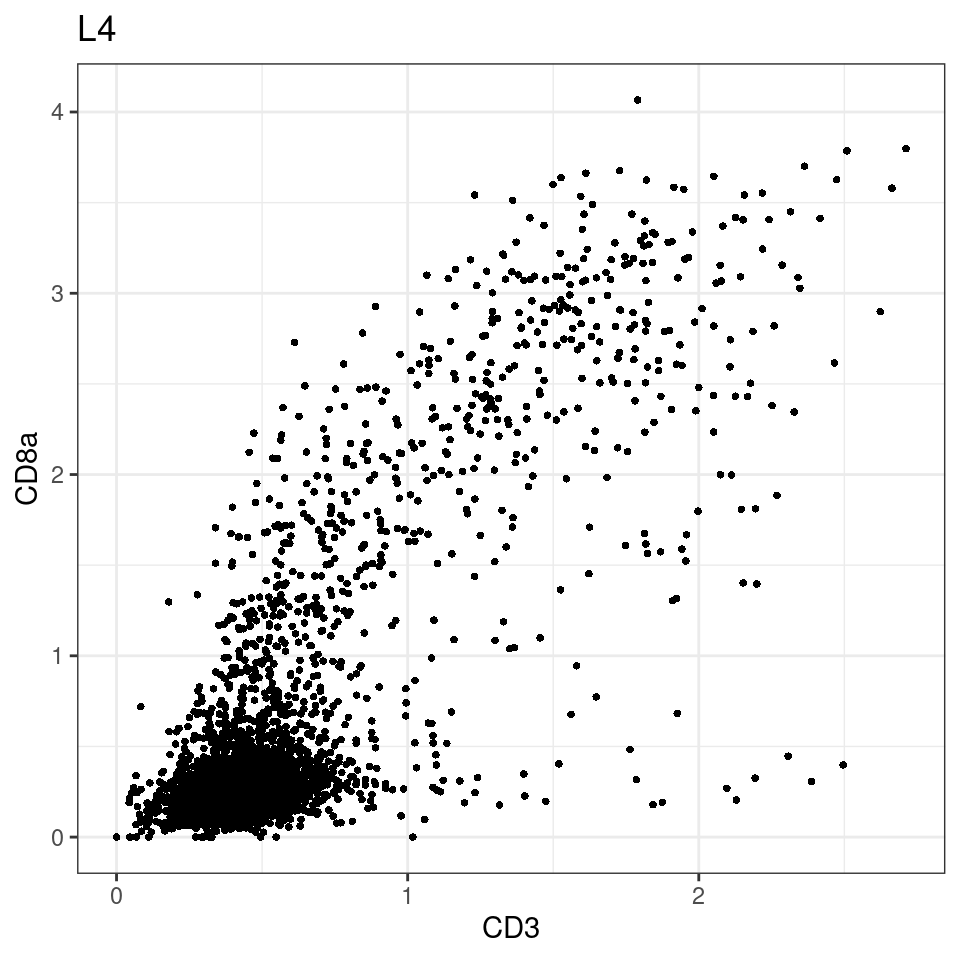
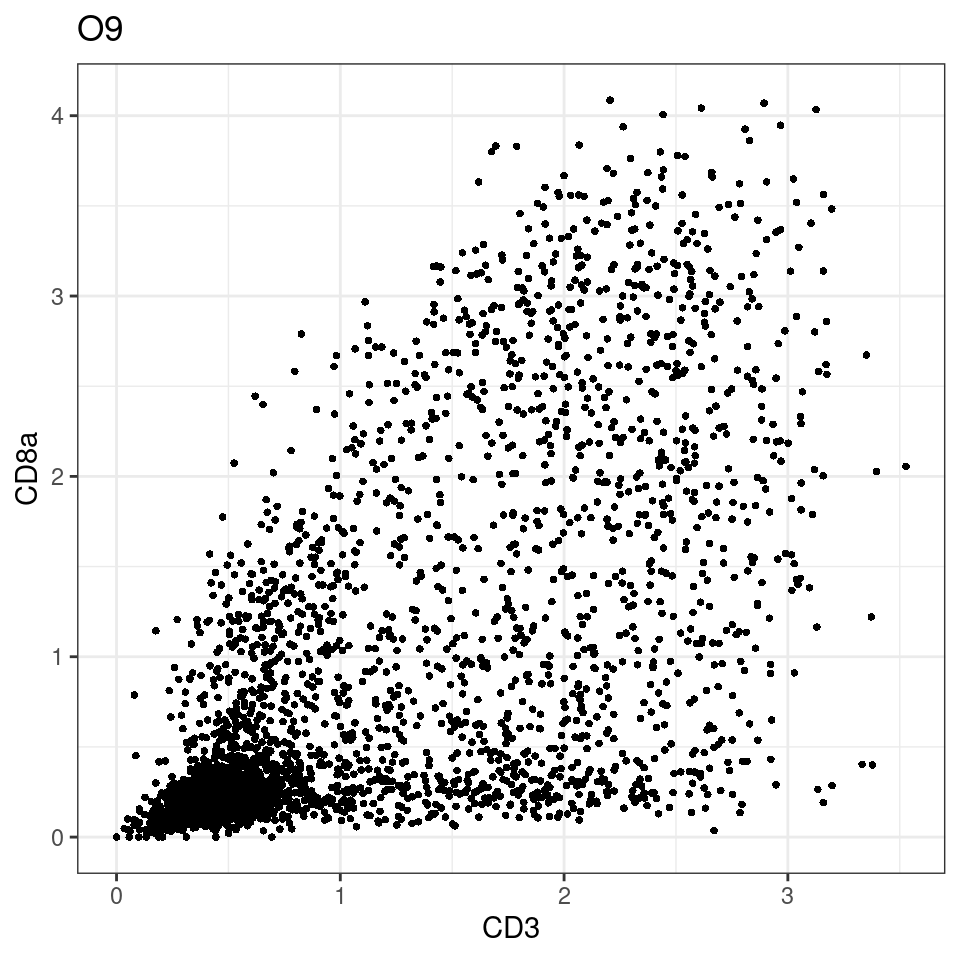
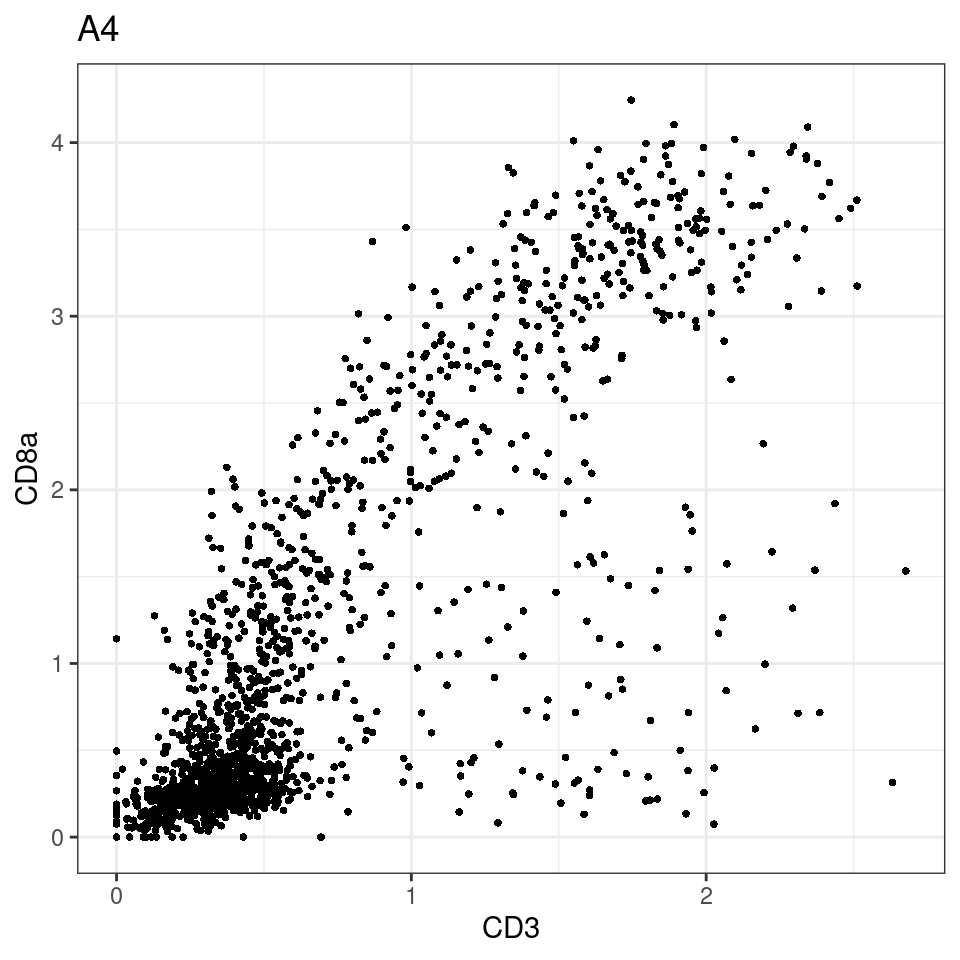
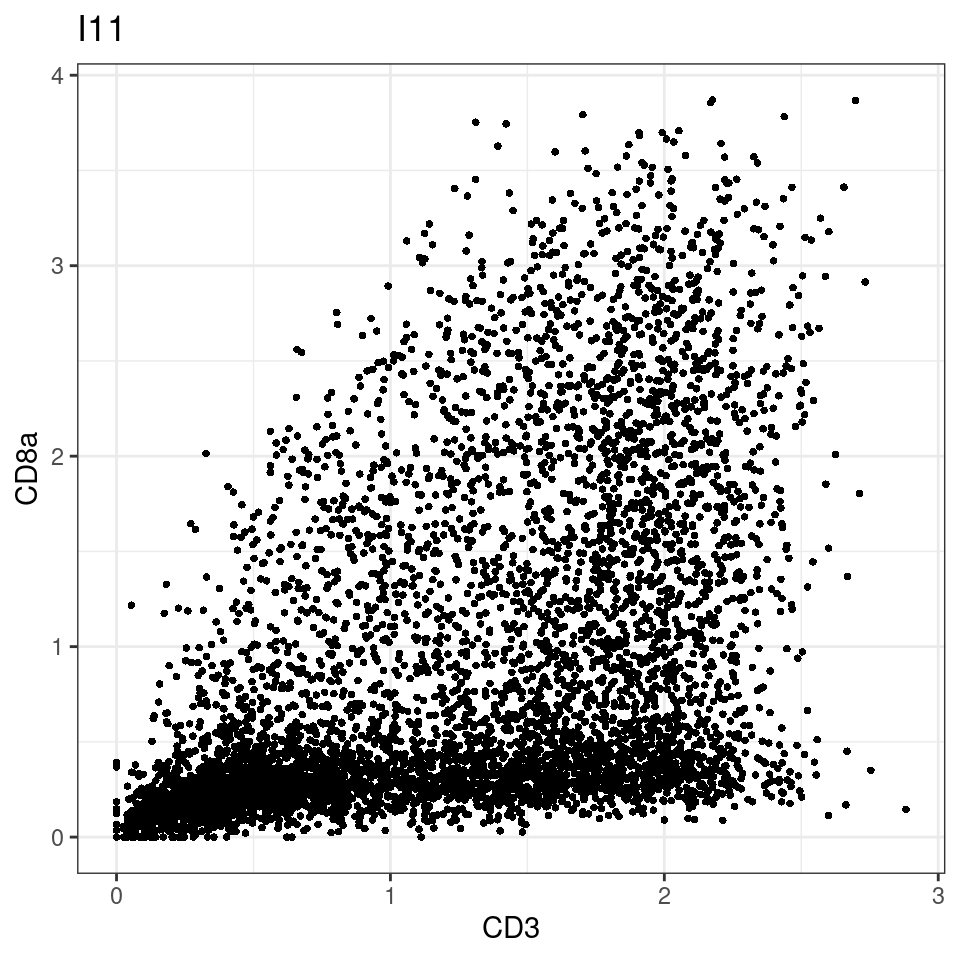
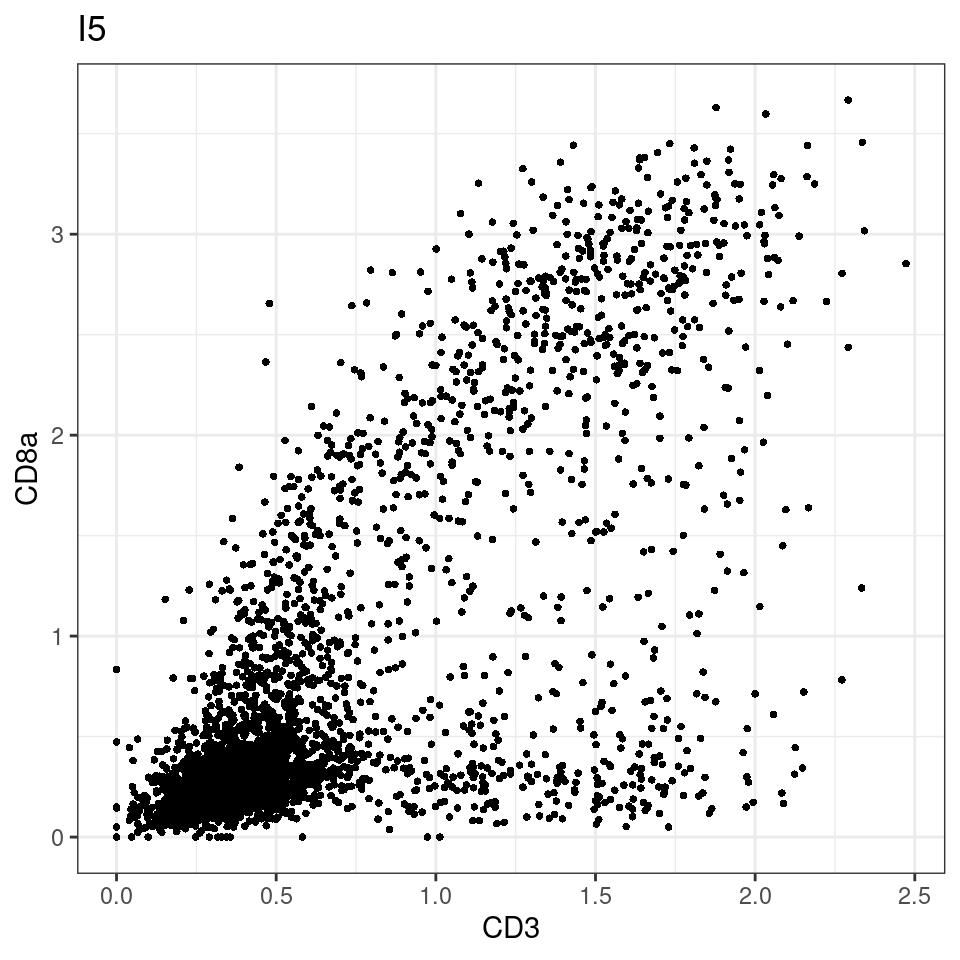
CD8 cut-off at asinh = 2
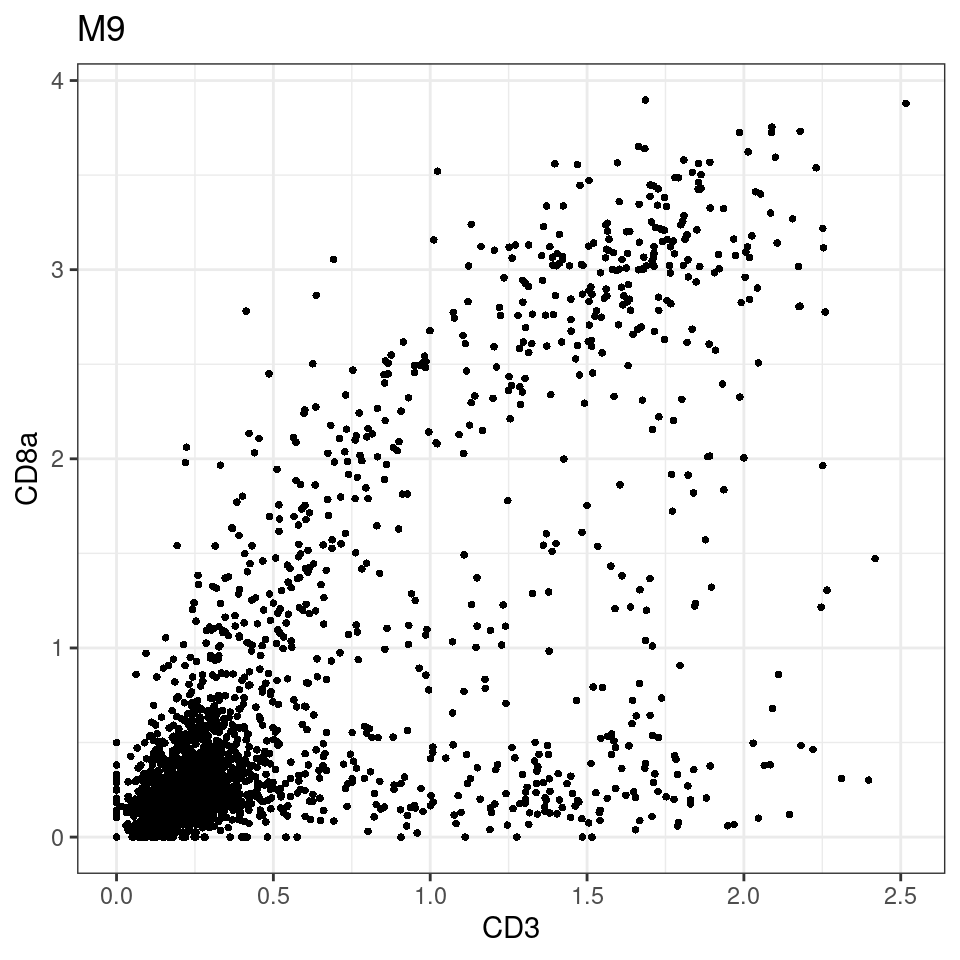
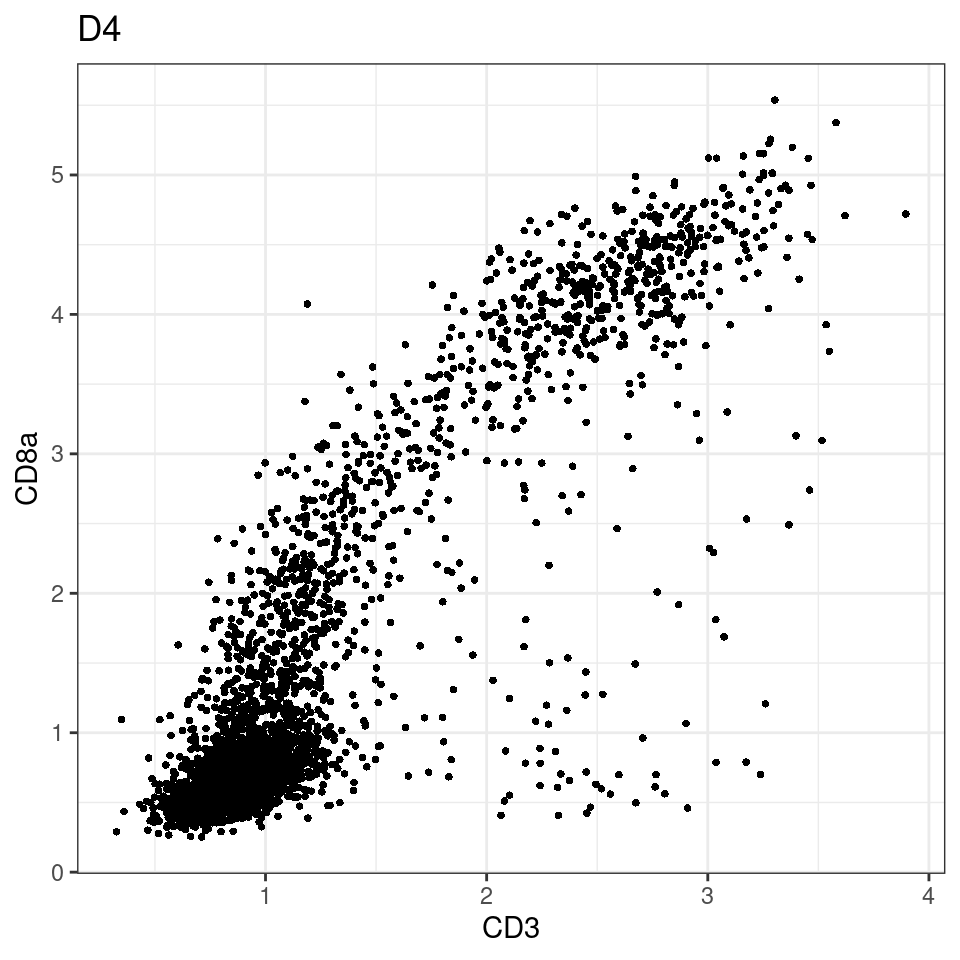
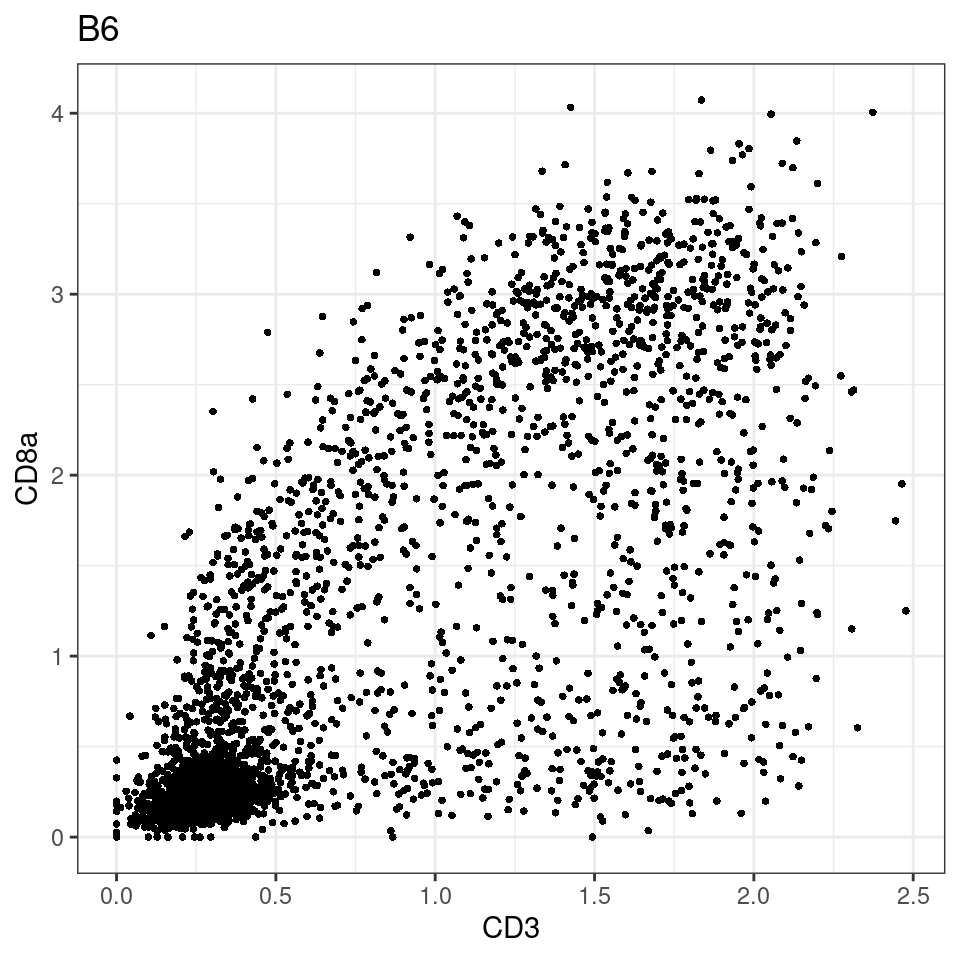
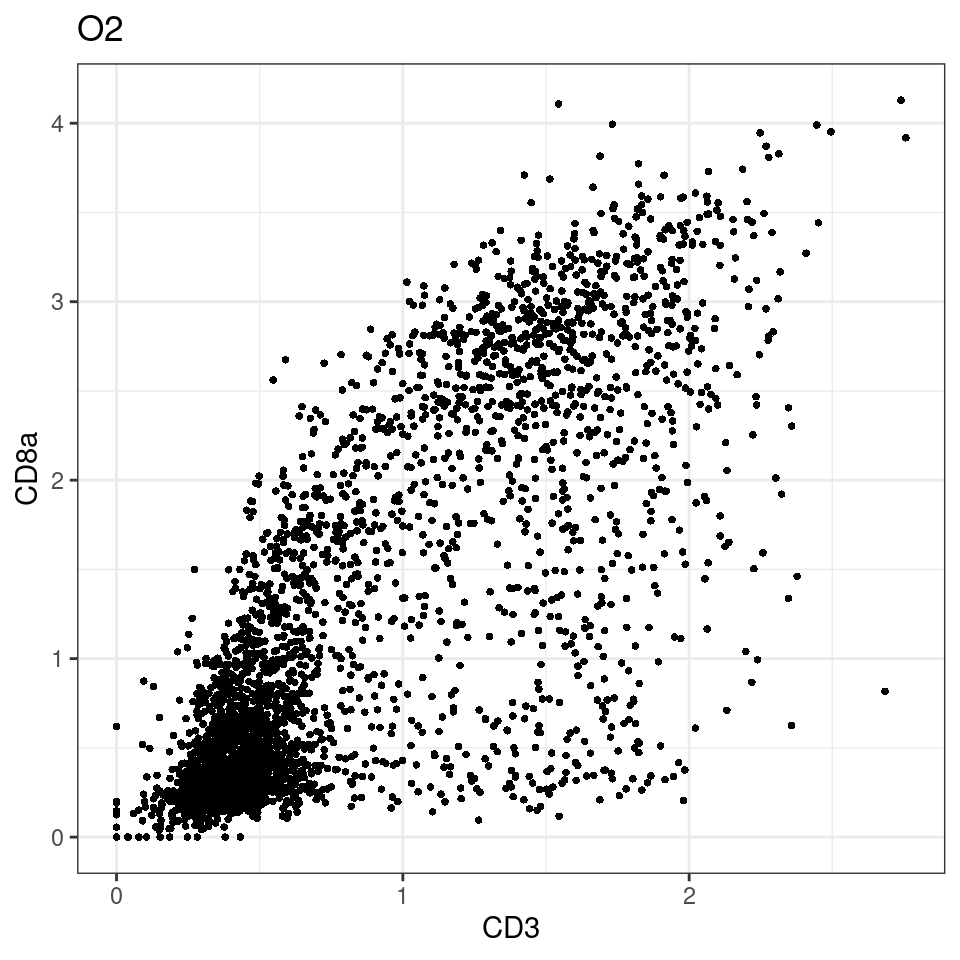
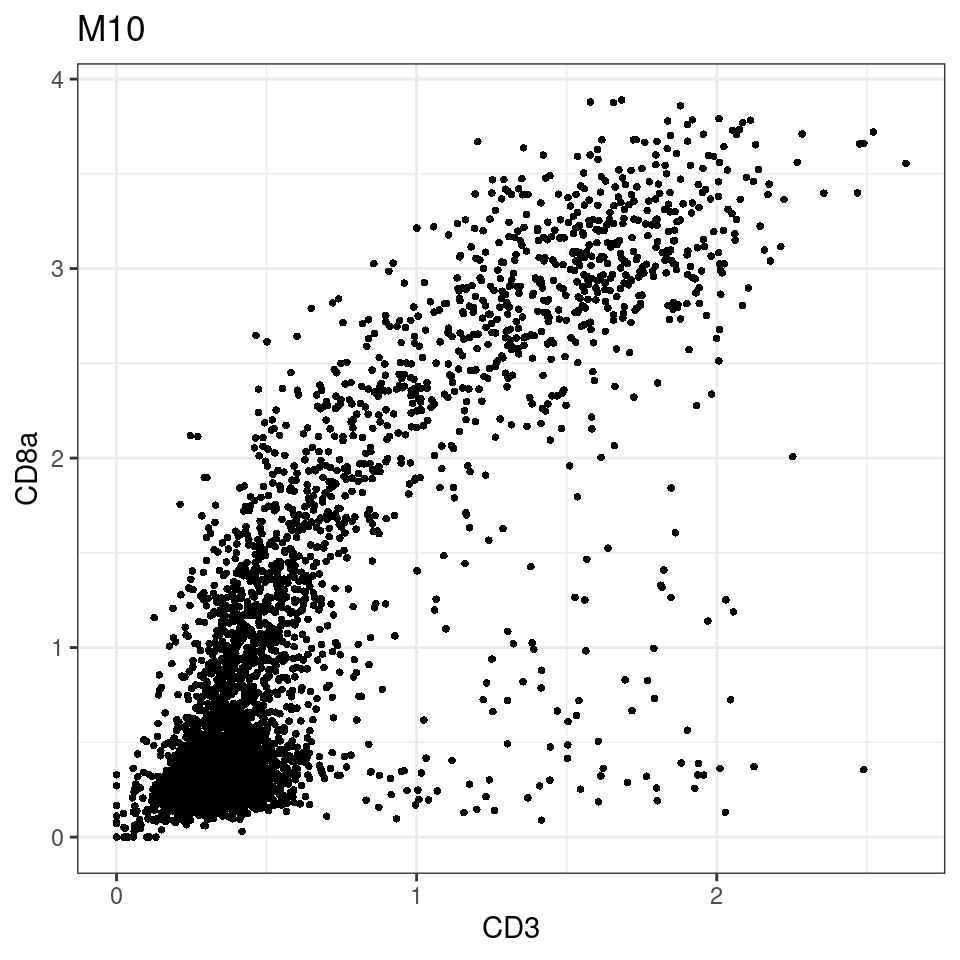
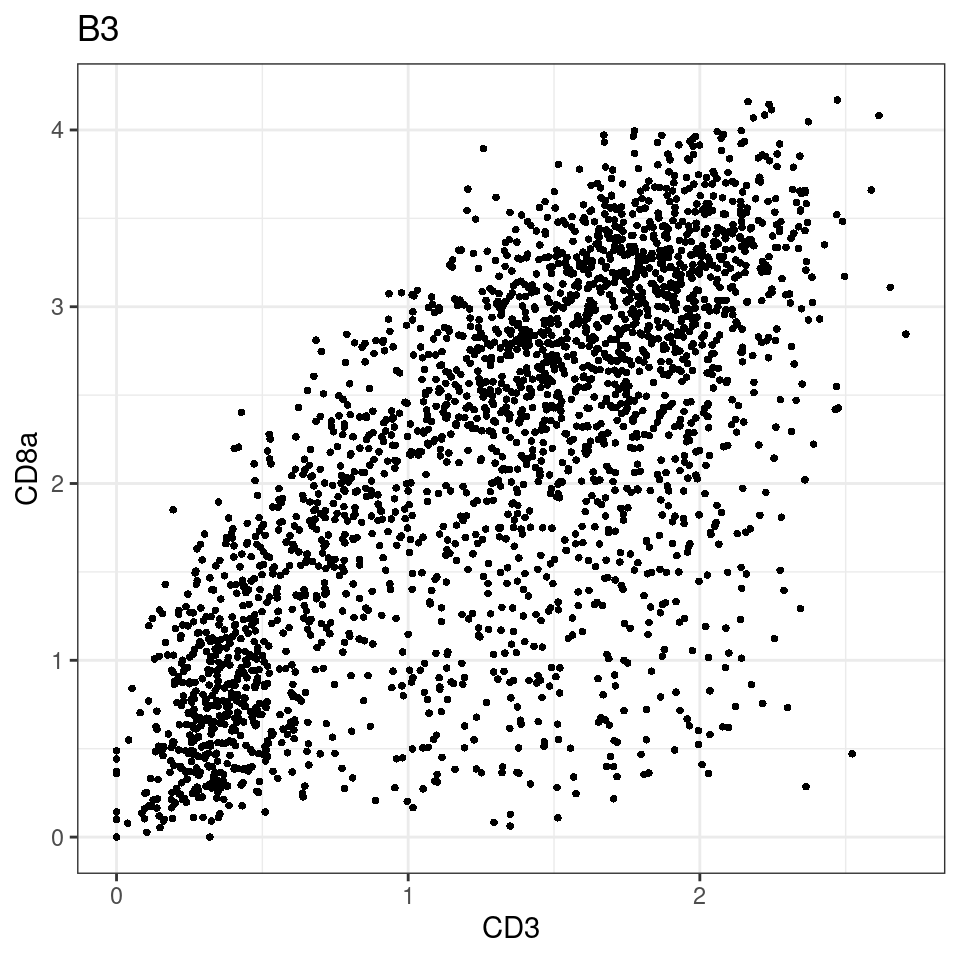
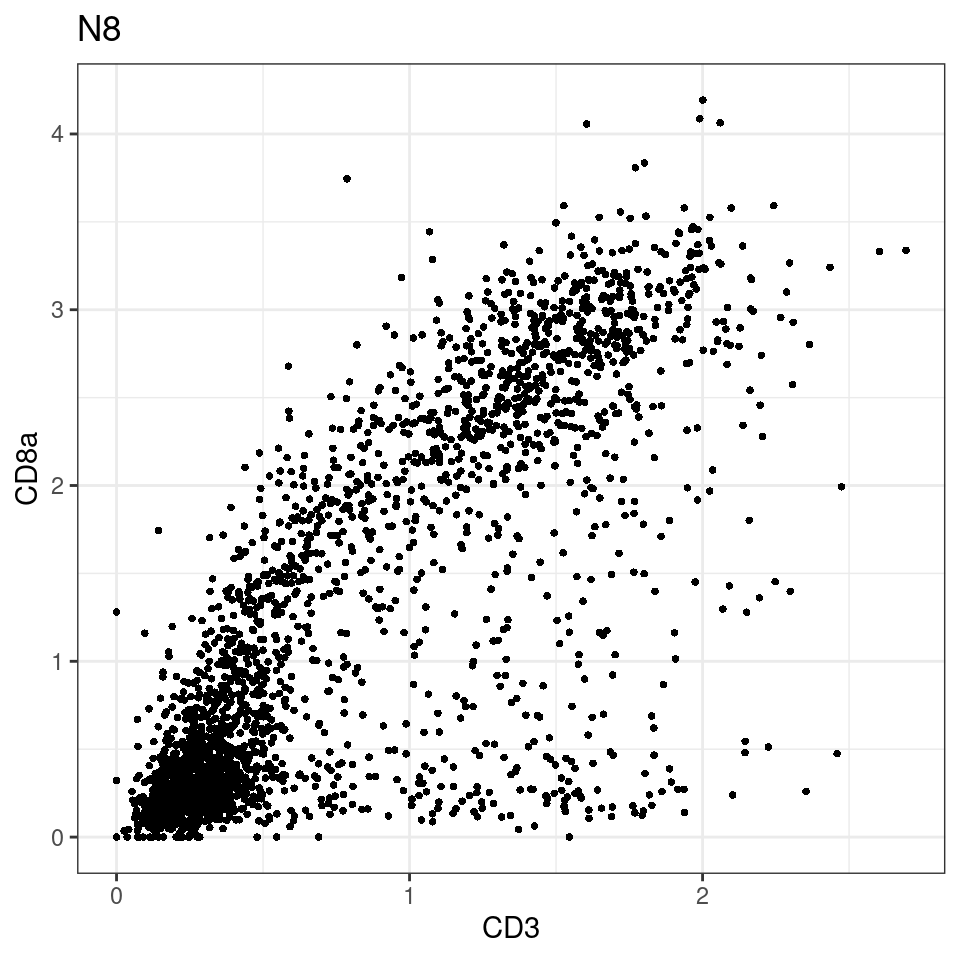
for (i in unique(sce$Description)) {
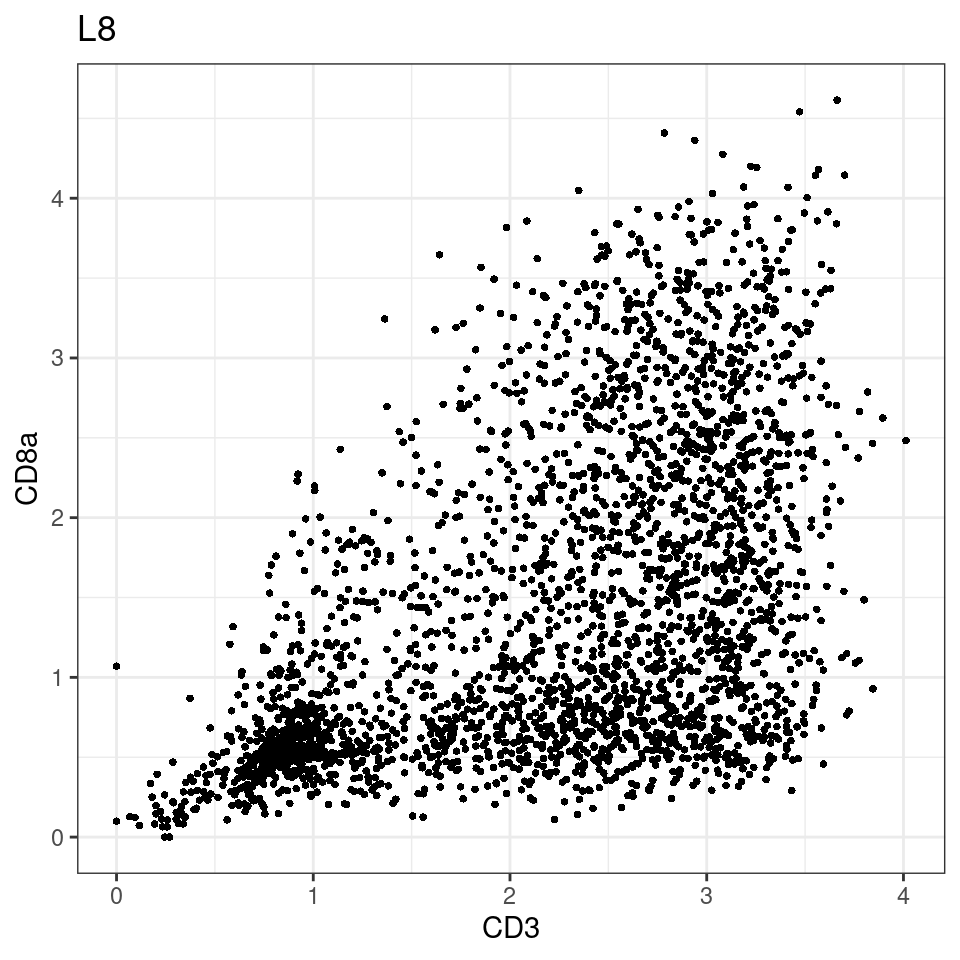
p <-dittoScatterPlot(sce[,which(sce$Description == i )],x.var = "CD3",y.var = "CD8a",assay.x = "asinh",assay.y = "asinh", main=i,)
plot(p)
}
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
| Version | Author | Date |
|---|---|---|
| 5418dcd | toobiwankenobi | 2022-02-22 |
general cut off for CD3: 1
Define Cell Types manually
sce$celltype <- "other"
sce$CXCL13 <- "negative"
sce[,which(t(assay(sce,"asinh"))[,"CD8a"] > 2 & t(assay(sce,"asinh"))[,"CD3"] > 1) ]$celltype <- "CD8_Tcell"
sce[,which(t(assay(sce,"asinh"))[,"CXCL13"] > 2 ) ]$CXCL13 <- "positive"
sce[,which(sce$celltype == "CD8_Tcell" & sce$CXCL13 == "positive")]$celltype <- "CD8_CXCL13+_Tcell"
sce[,which(sce$celltype == "other" & sce$CXCL13 == "positive")]$celltype <- "other_CXCL13+"Save SCE object
saveRDS(sce, file = "data/data_for_analysis/sce_dysfunction.rds")
sessionInfo()R version 4.1.2 (2021-11-01)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.3 LTS
Matrix products: default
BLAS/LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.8.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] scran_1.22.1 ggpubr_0.4.0
[3] forcats_0.5.1 stringr_1.4.0
[5] dplyr_1.0.7 purrr_0.3.4
[7] readr_2.1.2 tidyr_1.2.0
[9] tibble_3.1.6 tidyverse_1.3.1
[11] Hmisc_4.6-0 Formula_1.2-4
[13] survival_3.2-13 lattice_0.20-45
[15] cowplot_1.1.1 scales_1.1.1
[17] scater_1.22.0 scuttle_1.4.0
[19] dittoSeq_1.6.0 ggplot2_3.3.5
[21] BiocNeighbors_1.12.0 BiocParallel_1.28.3
[23] imcRtools_1.0.2 SpatialExperiment_1.4.0
[25] SingleCellExperiment_1.16.0 SummarizedExperiment_1.24.0
[27] Biobase_2.54.0 GenomicRanges_1.46.1
[29] GenomeInfoDb_1.30.1 IRanges_2.28.0
[31] S4Vectors_0.32.3 BiocGenerics_0.40.0
[33] MatrixGenerics_1.6.0 matrixStats_0.61.0
[35] workflowr_1.7.0
loaded via a namespace (and not attached):
[1] R.methodsS3_1.8.1 bit64_4.0.5
[3] knitr_1.37 irlba_2.3.5
[5] DelayedArray_0.20.0 R.utils_2.11.0
[7] data.table_1.14.2 rpart_4.1.16
[9] RCurl_1.98-1.5 generics_0.1.2
[11] ScaledMatrix_1.2.0 callr_3.7.0
[13] terra_1.5-17 proxy_0.4-26
[15] bit_4.0.4 tzdb_0.2.0
[17] xml2_1.3.3 lubridate_1.8.0
[19] httpuv_1.6.5 assertthat_0.2.1
[21] viridis_0.6.2 xfun_0.29
[23] hms_1.1.1 jquerylib_0.1.4
[25] evaluate_0.14 promises_1.2.0.1
[27] fansi_1.0.2 dbplyr_2.1.1
[29] readxl_1.3.1 igraph_1.2.11
[31] DBI_1.1.2 htmlwidgets_1.5.4
[33] ellipsis_0.3.2 backports_1.4.1
[35] svgPanZoom_0.3.4 sparseMatrixStats_1.6.0
[37] vctrs_0.3.8 abind_1.4-5
[39] withr_2.4.3 ggforce_0.3.3
[41] cytomapper_1.6.0 checkmate_2.0.0
[43] vroom_1.5.7 svglite_2.0.0
[45] cluster_2.1.2 crayon_1.4.2
[47] labeling_0.4.2 edgeR_3.36.0
[49] pkgconfig_2.0.3 units_0.7-2
[51] tweenr_1.0.2 vipor_0.4.5
[53] nnet_7.3-17 rlang_1.0.0
[55] lifecycle_1.0.1 modelr_0.1.8
[57] rsvd_1.0.5 cellranger_1.1.0
[59] rprojroot_2.0.2 polyclip_1.10-0
[61] tiff_0.1-11 Matrix_1.4-0
[63] raster_3.5-15 carData_3.0-5
[65] Rhdf5lib_1.16.0 reprex_2.0.1
[67] base64enc_0.1-3 beeswarm_0.4.0
[69] RTriangle_1.6-0.10 whisker_0.4
[71] ggridges_0.5.3 processx_3.5.2
[73] pheatmap_1.0.12 png_0.1-7
[75] viridisLite_0.4.0 rjson_0.2.21
[77] bitops_1.0-7 shinydashboard_0.7.2
[79] getPass_0.2-2 R.oo_1.24.0
[81] KernSmooth_2.23-20 rhdf5filters_1.6.0
[83] DelayedMatrixStats_1.16.0 classInt_0.4-3
[85] jpeg_0.1-9 rstatix_0.7.0
[87] ggsignif_0.6.3 beachmat_2.10.0
[89] magrittr_2.0.2 plyr_1.8.6
[91] zlibbioc_1.40.0 compiler_4.1.2
[93] dqrng_0.3.0 concaveman_1.1.0
[95] RColorBrewer_1.1-2 cli_3.1.1
[97] XVector_0.34.0 ps_1.6.0
[99] htmlTable_2.4.0 MASS_7.3-55
[101] tidyselect_1.1.1 stringi_1.7.6
[103] highr_0.9 yaml_2.2.2
[105] BiocSingular_1.10.0 locfit_1.5-9.4
[107] latticeExtra_0.6-29 ggrepel_0.9.1
[109] grid_4.1.2 sass_0.4.0
[111] EBImage_4.36.0 tools_4.1.2
[113] parallel_4.1.2 rstudioapi_0.13
[115] bluster_1.4.0 foreign_0.8-82
[117] git2r_0.29.0 metapod_1.2.0
[119] gridExtra_2.3 farver_2.1.0
[121] ggraph_2.0.5 DropletUtils_1.14.2
[123] digest_0.6.29 shiny_1.7.1
[125] Rcpp_1.0.8 car_3.0-12
[127] broom_0.7.12 later_1.3.0
[129] httr_1.4.2 sf_1.0-5
[131] colorspace_2.0-2 rvest_1.0.2
[133] fs_1.5.2 splines_4.1.2
[135] statmod_1.4.36 graphlayouts_0.8.0
[137] sp_1.4-6 systemfonts_1.0.3
[139] xtable_1.8-4 jsonlite_1.7.3
[141] tidygraph_1.2.0 R6_2.5.1
[143] pillar_1.7.0 htmltools_0.5.2
[145] mime_0.12 glue_1.6.1
[147] fastmap_1.1.0 DT_0.20
[149] fftwtools_0.9-11 class_7.3-20
[151] codetools_0.2-18 utf8_1.2.2
[153] bslib_0.3.1 ggbeeswarm_0.6.0
[155] magick_2.7.3 limma_3.50.0
[157] rmarkdown_2.11 munsell_0.5.0
[159] e1071_1.7-9 rhdf5_2.38.0
[161] GenomeInfoDbData_1.2.7 HDF5Array_1.22.1
[163] haven_2.4.3 gtable_0.3.0