Orthology inference: How are genes related across Schistocerca species?
Maeva Techer
2025-02-10
Last updated: 2025-02-10
Checks: 5 2
Knit directory:
locust-comparative-genomics/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221025) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Schistocerca/Results_I2/ | data/orthofinder/Schistocerca/Results_I2 |
| /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/ | data/orthofinder/Polyneoptera/Results_I2 |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 34c299a. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: data/.DS_Store
Ignored: data/DEG-results/.DS_Store
Ignored: data/WGCNA_input/.DS_Store
Ignored: data/WGCNA_output/.DS_Store
Ignored: data/behavioral_data/.DS_Store
Ignored: data/behavioral_data/Raw_data/.DS_Store
Ignored: data/list/.DS_Store
Ignored: data/list/GO_Annotations/.DS_Store
Ignored: data/orthofinder/.DS_Store
Ignored: data/orthofinder/Polyneoptera/.DS_Store
Ignored: data/orthofinder/Polyneoptera/Results_I2/.DS_Store
Ignored: data/orthofinder/Polyneoptera/Results_I2/Orthogroups/.DS_Store
Ignored: data/orthofinder/Polyneoptera/Results_I5/.DS_Store
Ignored: data/orthofinder/Polyneoptera/Results_I5/Orthogroups/.DS_Store
Ignored: data/orthofinder/Schistocerca/.DS_Store
Ignored: data/orthofinder/Schistocerca/Results_I2/.DS_Store
Ignored: data/orthofinder/Schistocerca/Results_I2/Orthogroups/.DS_Store
Ignored: data/orthofinder/Schistocerca/Results_I5/.DS_Store
Ignored: data/orthofinder/Schistocerca/Results_I5/Orthogroups/.DS_Store
Ignored: data/overlap/.DS_Store
Ignored: data/readcounts/.DS_Store
Untracked files:
Untracked: analysis/3_compiling_tables.Rmd
Untracked: data/orthofinder/Polyneoptera/Results_I5/Orthogroups/Orthogroups.tsv
Untracked: data/orthofinder/Polyneoptera/Results_I5/Orthogroups/Orthogroups.txt
Untracked: data/orthofinder/Polyneoptera/Results_I5/Orthogroups/Orthogroups_reprocessed.tsv
Untracked: data/orthofinder/Polyneoptera/Results_I5/Orthogroups/Orthogroups_reprocessed.txt
Untracked: data/orthofinder/Schistocerca/Results_I2/Orthogroups/Orthogroups.tsv
Untracked: data/orthofinder/Schistocerca/Results_I2/Orthogroups/Orthogroups.txt
Untracked: data/orthofinder/Schistocerca/Results_I2/Orthogroups/Orthogroups_reprocessed.tsv
Untracked: data/orthofinder/Schistocerca/Results_I2/Orthogroups/Orthogroups_reprocessed.txt
Unstaged changes:
Modified: analysis/2_orthologs-prediction.Rmd
Modified: data/orthofinder/Polyneoptera/Results_I2/Orthogroups_13species_Jan2025.txt
Modified: data/orthofinder/Polyneoptera/Results_I2/Orthogroups_genesprotein_Schisto_Jan2025.txt
Modified: data/orthofinder/Polyneoptera/Results_I2/Orthogroups_genesproteinbiotype_13species_Jan2025.csv
Modified: data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_A. simplex.pdf
Modified: data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_B. rossius.pdf
Modified: data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_C. secundus.pdf
Modified: data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_D. australis.pdf
Modified: data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_G. bimaculatus.pdf
Modified: data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_G. longicornis.pdf
Modified: data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_P. americana.pdf
Modified: data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_americana.pdf
Modified: data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_cancellata.pdf
Modified: data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_cubense.pdf
Modified: data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_gregaria.pdf
Modified: data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_nitens.pdf
Modified: data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_piceifrons.pdf
Modified: data/orthofinder/Polyneoptera/Results_I2/SingleCopyOrthogroups_genesprotein_13species_Jan2025.txt
Modified: data/orthofinder/Schistocerca/Results_I2/Orthogroups_Schistocerca_Jan2025.txt
Modified: data/orthofinder/Schistocerca/Results_I2/Orthogroups_genesproteinbiotype_Schistocerca_Jan2025.csv
Modified: data/orthofinder/Schistocerca/Results_I2/Plots_Schistocerca/VerticalStackedBar_americana.pdf
Modified: data/orthofinder/Schistocerca/Results_I2/Plots_Schistocerca/VerticalStackedBar_cancellata.pdf
Modified: data/orthofinder/Schistocerca/Results_I2/Plots_Schistocerca/VerticalStackedBar_cubense.pdf
Modified: data/orthofinder/Schistocerca/Results_I2/Plots_Schistocerca/VerticalStackedBar_gregaria.pdf
Modified: data/orthofinder/Schistocerca/Results_I2/Plots_Schistocerca/VerticalStackedBar_nitens.pdf
Modified: data/orthofinder/Schistocerca/Results_I2/Plots_Schistocerca/VerticalStackedBar_piceifrons.pdf
Modified: data/orthofinder/Schistocerca/Results_I2/SingleCopyOrthogroups_genesprotein_6species_Jan2025.txt
Deleted: data/overlap/summary_DEGs_Orthogroups_togregaria.csv
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown
(analysis/2_orthologs-prediction.Rmd) and HTML
(docs/2_orthologs-prediction.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | db8b525 | Maeva TECHER | 2025-02-06 | update overlap |
| html | db8b525 | Maeva TECHER | 2025-02-06 | update overlap |
| Rmd | e55bac6 | Maeva TECHER | 2025-01-26 | Updating the github |
| html | e55bac6 | Maeva TECHER | 2025-01-26 | Updating the github |
| Rmd | faf2db3 | Maeva TECHER | 2025-01-13 | update markdown |
| html | faf2db3 | Maeva TECHER | 2025-01-13 | update markdown |
| Rmd | b80db34 | Maeva TECHER | 2025-01-13 | Adding selection analysis part |
| html | b80db34 | Maeva TECHER | 2025-01-13 | Adding selection analysis part |
| Rmd | 3fa8e62 | Maeva TECHER | 2024-11-09 | updated analysis |
| html | 3fa8e62 | Maeva TECHER | 2024-11-09 | updated analysis |
| Rmd | edb70fe | Maeva TECHER | 2024-11-08 | overlap and deg results created |
| html | edb70fe | Maeva TECHER | 2024-11-08 | overlap and deg results created |
| html | 7f1d1fe | Maeva TECHER | 2024-11-01 | Build site. |
| Rmd | f01f1cf | Maeva TECHER | 2024-11-01 | Adding new files and docs |
| html | f01f1cf | Maeva TECHER | 2024-11-01 | Adding new files and docs |
| html | ba35b82 | Maeva A. TECHER | 2024-06-20 | Build site. |
| html | c006e71 | Maeva A. TECHER | 2024-06-20 | change |
| html | 82ef59f | Maeva A. TECHER | 2024-05-16 | Build site. |
| Rmd | 151afb3 | Maeva A. TECHER | 2024-05-16 | wflow_publish("analysis/2_orthologs-prediction.Rmd") |
| html | 4dd0e26 | Maeva A. TECHER | 2024-05-15 | Build site. |
| Rmd | 38ef822 | Maeva A. TECHER | 2024-05-15 | wflow_publish("analysis/2_orthologs-prediction.Rmd") |
| html | ce82fe8 | Maeva A. TECHER | 2024-05-14 | Build site. |
| Rmd | 3ca5aee | Maeva A. TECHER | 2024-05-14 | wflow_publish("analysis/2_orthologs-prediction.Rmd") |
| html | ac06f45 | Maeva A. TECHER | 2024-05-14 | Build site. |
| Rmd | be09a11 | Maeva A. TECHER | 2024-05-14 | update markdown |
| html | be09a11 | Maeva A. TECHER | 2024-05-14 | update markdown |
| html | 0837617 | Maeva A. TECHER | 2024-01-30 | Build site. |
| html | f701a01 | Maeva A. TECHER | 2024-01-30 | reupdate |
| html | 2ca8696 | Maeva A. TECHER | 2024-01-30 | Build site. |
| html | 5c5487e | Maeva A. TECHER | 2024-01-30 | Build site. |
| html | 0135a6e | Maeva A. TECHER | 2024-01-30 | Build site. |
| Rmd | 505a8dc | Maeva A. TECHER | 2024-01-30 | wflow_publish("analysis/2_orthologs-prediction.Rmd") |
| html | 579056a | Maeva A. TECHER | 2024-01-30 | Build site. |
| Rmd | f50a3dd | Maeva A. TECHER | 2024-01-30 | wflow_publish("analysis/2_orthologs-prediction.Rmd") |
| html | f831779 | Maeva A. TECHER | 2024-01-24 | Build site. |
| html | 1b09cbe | Maeva A. TECHER | 2024-01-24 | remove |
| html | 79006db | Maeva A. TECHER | 2024-01-24 | Build site. |
| Rmd | b195f57 | Maeva A. TECHER | 2024-01-24 | refresh |
| html | b195f57 | Maeva A. TECHER | 2024-01-24 | refresh |
| html | dfc68c7 | Maeva A. TECHER | 2023-12-18 | Build site. |
| Rmd | 53877fa | Maeva A. TECHER | 2023-12-18 | add pages |
Comparative genomics and ortholog genes with OrthoFinder
We wanted to compare the six genomes of Schistocerca to get insights on gene evolution and relationships regarding their numbers, content, function and location. In order to achieve this, we need to identify groups of orthologous genes among our species of interest, considering at least one outgroup.
Orthologs are genes from different species that originated from a single ancestral gene and evolved through speciation events. However since genes can be lost or duplicated during evolution, some genes may not have exactly one orthologue in the genome of another species. Here we will separate the 1:1 orthologs to the the concept of orthogroups. Orthogroups can contain 1:1 orthologs but also several several orthologs from different species, including paralogs and one-to-many orthologs. Paralogs are genes within the same species that have originated from a shared ancestral genes but have diverged over time following gene duplication events.
Note:We used OrthoFinder to identify the orthogroups using amino acid sequences from the longest isoform of each gene. For this part, refers to the well curated pipeline FormicidaeMolecularEvolution by Megan Barkdull (Assistant Curator of Entomology at the Natural History Museum of Los Angeles County). We describe below the modifications made and mostly copied the workflow from her Github.
1. Downloading data
We created the file “input-XXX.txt” as described to automatically download the coding sequence, protein sequence, GFF annotation data for each of our six Schistocerca species and annotated outgroups. The outgroups were choosen as close phylogenetic species with a RefSeq genome showing 1) a chromosome length, 2) associated with an annotation, 3) large size and 4) hybrid techniques used for assembly (last NCBI search: 28 April 2024).
Our target species for this study:
* The desert locust Schistocerca
gregaria
* The South American locust Schistocerca
cancellata
* The Central American locust Schistocerca
piceifrons
* The American grasshopper Schistocerca
americana
* The bird grasshopper Schistocerca
serialis cubense
* The vagrant locust Schistocerca
nitens
Our Polyneoptera outgroup species:
* The Mormon cricket Anabrus
simplex. Reason: Orthoptera close relative available
with chromosome length.
* The long cercus field cricket Gryllus
longicercus. Reason: Orthoptera close relative available
with scaffold length and a user submitted annotation.
* The two-spotted cricket Gryllus
bimaculatus. Reason: Orthoptera close relative available
with chromosome length.
* The Lord Howe Island stick insect Dryococelus
australis. Reason: Polyneoptera close relative available
with chromosome length.
* The European stick insect Bacillus
rossius redtenbacheri. Reason: Polyneoptera close
relative available with chromosome length.
* The drywood termite Cryptotermes
secundus. Reason: Eusocial insect with caste
determination phenotypic plasticity.
* The American cockroach Periplaneta
americana. Reason: Polyneoptera close relative available
with chromosome length.
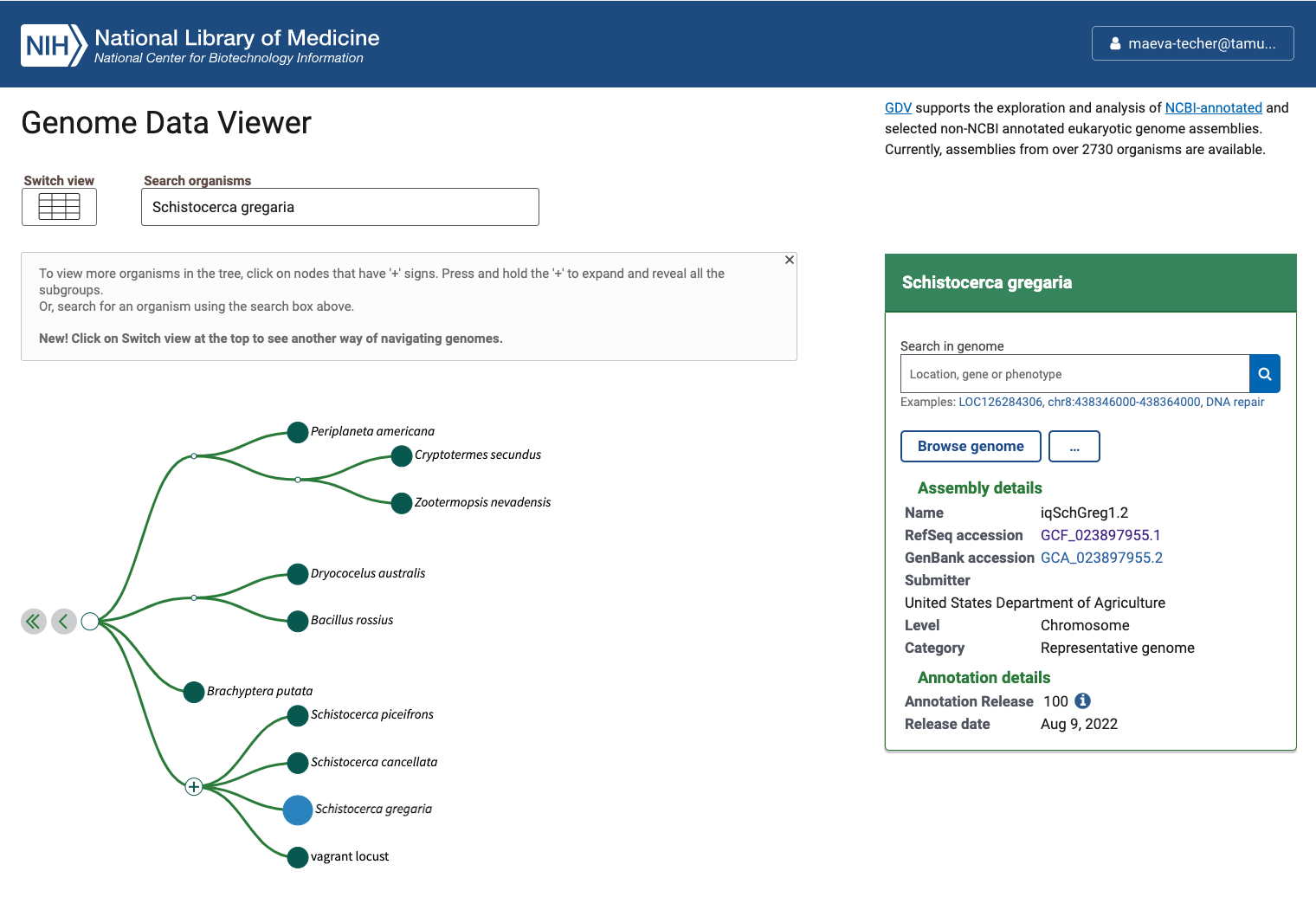
Status of Genome Data Viewer on NCBI for selecting our
outgroups
NB: During our search, we identified very valuable genomes but for which no annotation was available (e.g., CAU_Lmig_1.0 for Locusta migratoria , iqMecThal1.2 Meconema thalassinum). However upon request to NCBI team, we were informed that the Locusta genome has not been annotated because of the presence of large bacterial contamination and Meconema does not possess RNA SRAs to help with the annotation.
Using the FTP NCBI link associated with each RefSeq, we created the input file.
./scripts/DataDownload ./scripts/inputurls_18polyneoptera_Nov2024.txt On the TAMU Grace cluster, when you download do not run it on a node
as it does not work. Run it from the login node or transfer the files
already preloaded. This will creates a folder ./1_RawData
with 3 files for each species.
In summary, below are the details of each RefSeq annotation that we will be using as input for OrthoFinder. It will be important to check that after steps 2-4, the number of protein coding genes OrthoFinder is similar to the initial input.
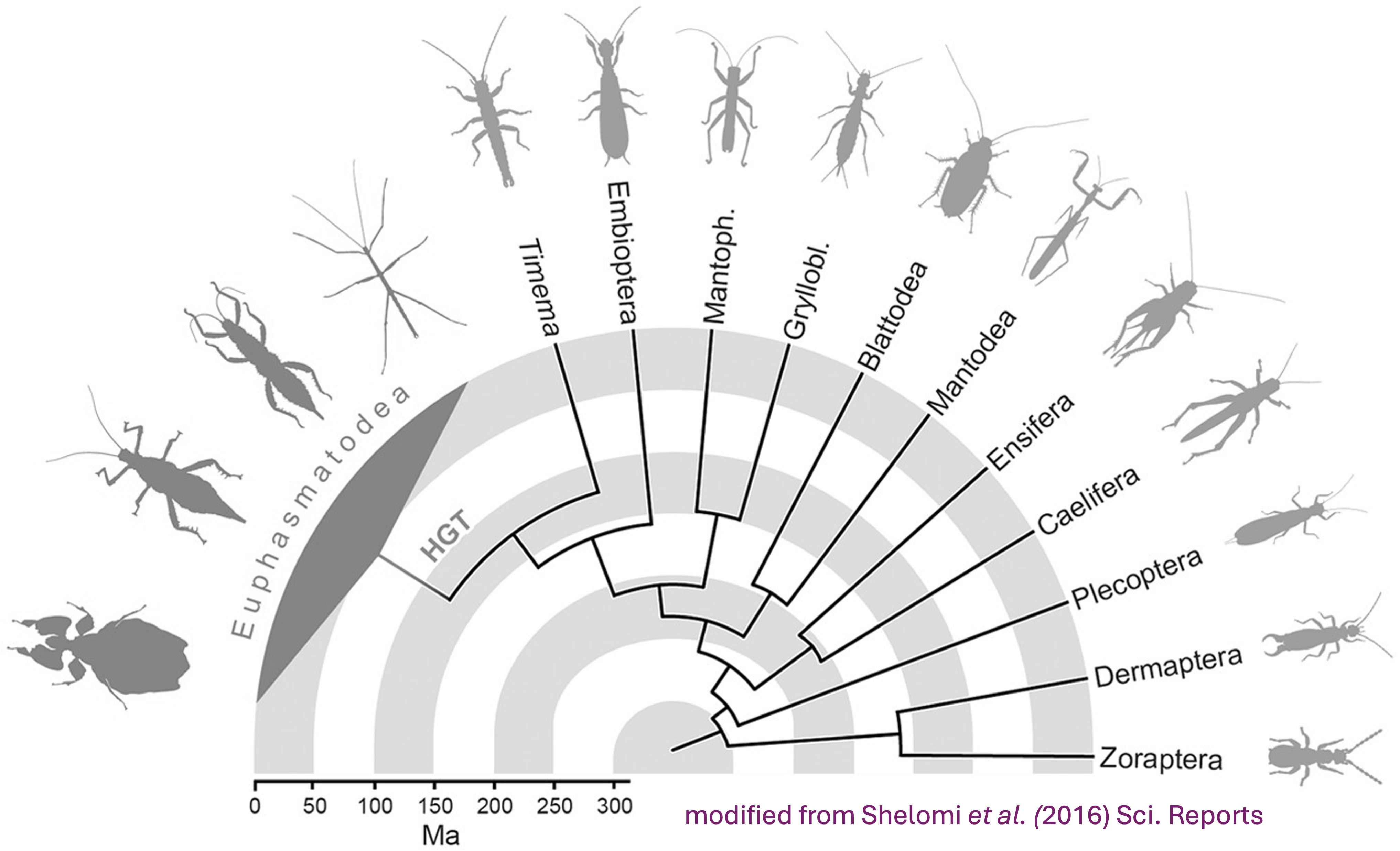
| Species | Order | Status | Genome_Size | Annotated_Genes | Protein_Coding |
|---|---|---|---|---|---|
| Schistocerca gregaria | Orthoptera | Locust | 8.7 Gb | 99467 | 19799 |
| Schistocerca cancellata | Orthoptera | Locust | 8.5 Gb | 103533 | 16907 |
| Schistocerca piceifrons | Orthoptera | Locust | 8.7 Gb | 96806 | 17490 |
| Schistocerca americana | Orthoptera | Grasshopper | 9.0 Gb | 81274 | 17662 |
| Schistocerca serialis cubense | Orthoptera | Grasshopper | 9.1 Gb | 75810 | 17237 |
| Schistocerca nitens | Orthoptera | Grasshopper | 8.8 Gb | 72560 | 17500 |
| Anabrus simplex (Idaho) | Orthoptera | Outgroup | 6.4 Gb | 27091 | 14866 |
| Gryllus bimaculatus | Orthoptera | Outgroup | 1.7 Gb | 17871 | NA |
| Gryllus longicercus | Orthoptera | Outgroup | 1.9 Gb | 14831 | NA |
| Bacillus rossius redtenbacheri | Phasmatodea | Outgroup | 1.6 Gb | 19298 | 14448 |
| Dryococelus australis | Phasmatodea | Outgroup | 3.4 Gb | 33793 | NA |
| Periplaneta americana | Blattodea | Outgroup | 3.1 Gb | 28416 | 28414 |
| Cryptotermes secundus | Blattodea | Outgroup | 3.1 Gb | 27047 | NA |
2. Selecting longest isoforms
Here again, we will follow the pipeline except that to run it on Grace cluster we will make small modifications. The idea is that we will use only the single longest isoform of each gene to ease the orthology analysis. While this might not always be the principal isoform of said gene, we will apply the same bias to all genes the same way.
To run R without bothering other users, we will claim one interactive node to make sure we can proactively update the package if there are some issues:
srun --ntasks 1 --cpus-per-task 4 --mem 10G --time 01:00:00 --pty bashThen we will use any package preloaded on the cluster before needed to install our own on our user library if needed. We will need Pandoc for loading orthologr
ml GCC/12.2.0 OpenMPI/4.1.4 R_tamu/4.3.1
export R_LIBS=$SCRATCH/R_LIBS_USER/
ml Pandoc/2.13We now simply run the script as indicated on the pipeline page:
./scripts/GeneRetrieval.R ./scripts/inputurls_13polyneoptera_Jan2025.txtIf the annotated genome is not done
by RefSeq but submitted by users:
When downloading genomes from NCBI, we found a few interesting ones that
were but annotated by users with a different format than NCBI Gnomon
pipeline. For example on the image below (when you zoom in), we can see
that the gene= field is present in RefSeq but is not in
users submitted but could be created using ID= field.
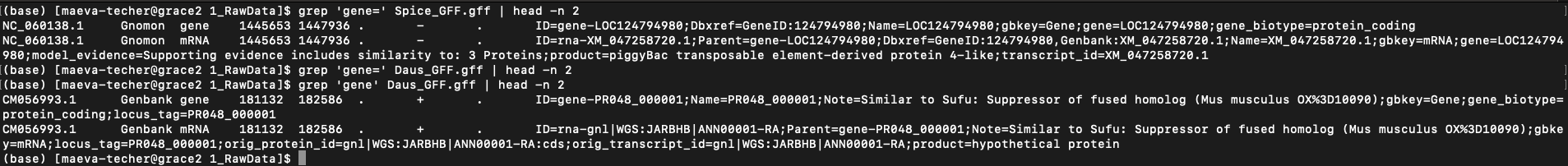
Example of difference in the presence of “gene” field between
S. piceifrons and Dryococelus australis
(3.4Gb).
To correct, I ran the following parsing code which append a new gene
column based on locus_tag:
# we need to modify the GFF, proteins and genome/transcript fasta file
sed -i 's/locus_tag=/gene=/g' {SPECIES}_GFF.gff
sed -i 's/locus_tag=/gene=/g' {SPECIES}_proteins.faa
sed -i 's/locus_tag=/gene=/g' {SPECIES}_transcripts.fasta
# then we check
grep 'gene=' {SPECIES}_GFF.gff | head -n 10We did it for Daus_GFF.gff, Dpunc_GFF.gff,
Gbima_GFF.gff, Glong_GFF.gff,
Pamer_GFF.gff and Tpodu_GFF.gff. Then instead
of running the GeneRetrieval.R, we computed manually (to check if error)
using the following R script:
library(orthologr)
library(tidyverse)
library(biomartr)
library(phylotools)
library(data.table)
# Define the species parameter
species <- "Gbima" # for example
#species <- "Glong"
#species <- "Daus"
#species <- "Dpunc"
#species <- "Pamer"
#species <- "Tpodu"
# Define file paths based on the species parameter
proteomeFile <- paste0("./1_RawData/", species, "_proteins.faa")
annotationFile <- paste0("./1_RawData/", species, "_GFF.gff")
longestIsoformsFile <- paste0("./2_LongestIsoforms/", species, "_longestIsoforms.fasta")
transcriptsFile <- paste0("./1_RawData/", species, "_transcripts.fasta")
filteredTranscriptsOutput <- paste0("./2_LongestIsoforms/", species, "_filteredTranscripts.fasta")
# Step 1: Retrieve the longest isoforms for Glong
retrieve_longest_isoforms(
proteome_file = proteomeFile,
annotation_file = annotationFile,
new_file = longestIsoformsFile,
annotation_format = "gff"
)
# Step 2: Load longest isoforms and transcript files
isoforms <- phylotools::read.fasta(longestIsoformsFile)
isoform_ids <- isoforms$seq.name # Get IDs of longest isoforms
isoform_ids
transcripts <- phylotools::read.fasta(transcriptsFile)
head(transcripts$seq.name)
# Step 3: Adjust headers in transcripts to match core identifiers
transcripts$seq.name <- str_extract(transcripts$seq.name, "(?<=cds_)[^ ]+") # Extract main identifier
transcripts$seq.name <- str_extract(transcripts$seq.name, "^[^_]+") # Keep only core ID without suffix
transcripts$seq.name
# Step 4: Filter transcripts to keep only those matching isoform IDs
filtered_transcripts <- transcripts %>%
filter(seq.name %in% isoform_ids)
# Step 5: Write the filtered transcripts to a new FASTA file with simplified headers
phylotools::dat2fasta(filtered_transcripts, outfile = filteredTranscriptsOutput)
message("Filtered transcripts for ", species, " saved to: ", filteredTranscriptsOutput)
Status of Gene Retrieval script when successful
| Species | Total_Peptides | Number_Kept_Isoforms |
|---|---|---|
| Schistocerca gregaria | 37988 | 19799 |
| Schistocerca cancellata | 26362 | 16907 |
| Schistocerca piceifrons | 25717 | 17490 |
| Schistocerca americana | 26125 | 17662 |
| Schistocerca serialis cubense | 27654 | 17237 |
| Schistocerca nitens | 28445 | 17500 |
| Anabrus simplex (Idaho) | X | X |
| Gryllus bimaculatus | 25032 | 17871 |
| Gryllus longicercus | 19656 | 14730 |
| Bacillus rossius redtenbacheri | 29758 | 14448 |
| Dryococelus australis | 33111 | 33111 |
| Periplaneta americana | 27047 | 27047 |
| Cryptotermes secundus | 29285 | 13170 |
3. Cleaning the raw data
For the cleaning step of the mbarkdull’s pipeline we simply followed the command line with no modifications.
./scripts/DataCleaning ./scripts/inputurls_13polyneoptera_Jan2025.txt 
Status of Data Cleaning script when successful
4. Translating nucleotide sequences to amino acid sequences
This step does not seems to be really
necessary because Orthofinder can immediately take the
XXX_longestIsoforms.fasta files. We just run the
scriptDataCleaningIsoforms_modif on it to rename and clean
it.:
As mentioned in the pipeline page, we will mostly need to use amino
acid sequences rather than protein and we need to translate the data we
downloaded. We will be using the Python script from mbarkdull
./scripts/TranscriptFilesTranslateScript.py but before that
we will modify our input file to only include only the 12 essential
species.
We changed the script in head lines and lines 25-27, to call the module directly from the cluster as follow:
#!/bin/bash
##NECESSARY JOB SPECIFICATIONS
#SBATCH --job-name=Transdecoder #Set the job name to "JobExample4"
#SBATCH --time=02:00:00 #Set the wall clock limit to 1hr and 30min
#SBATCH --ntasks=2 #Request 2 task
#SBATCH --cpus-per-task=4 #Request 8 task
#SBATCH --mem=20G #Request 50GB per node
ml GCC/10.2.0 OpenMPI/4.0.5 TransDecoder/5.5.0 Biopython/1.78 # Now we can run Transdecoder on the cleaned file:
echo "First, attempting TransDecoder run on $cleanName"
ml GCC/10.2.0 OpenMPI/4.0.5 TransDecoder/5.5.0
TransDecoder.LongOrfs -t $cleanName
TransDecoder.Predict -t $cleanName --single_best_onlyand then we launched the script by changing the top lines to put in sbatch:
sbatch ./scripts/DataTranslating_modif ./scripts/inputurls_13polyneoptera_Jan2025.txt 5. Running Orthofinder
Now we will finally run Orthofinder to identify groups of orthologous genones in our translated amino acid sequences. We will also use MAFFT to produce multiple sequence alignment across our six species of Schistocerca and the outgroups.
Instead of using the ./scripts/DataOrthofinder from the
pipeline, we will be running our own command line here. Before doing so,
we did manually the creating of folders and moving of fasta files as
follow:
mkdir LocustsGenomeEvolution/tmp
mkdir -p ./5_OrthoFinder/fasta
cp ./4_1_TranslatedData/OutputFiles/translated* ./5_OrthoFinder/fasta
cp ./3_CleanedData/*longestIsoforms.fasta ./5_OrthoFinder/fasta
cd ./5_OrthoFinder/fasta
rename translated '' translated*
# Loop through files and rename them
for file in cleaned*_longestIsoforms.fasta; do
# Remove "cleaned" and replace "_longestIsoforms" with "_filteredproteome"
new_name=$(echo "$file" | sed 's/^cleaned//; s/_longestIsoforms/_filteredproteome/')
mv "$file" "$new_name"
done
cd ../You can also rename them with a simple name like “S_americana.fasta”, this will help ease the visualization later on.
To run orthofinder onto our Grace cluster, I checked the
compatibility of the modules required and these are the versions
acceptable to run in sbatchorthofinder.sh:
#!/bin/bash
##NECESSARY JOB SPECIFICATIONS
#SBATCH --job-name=orthofinder-blast #Set the job name to "JobExample4"
#SBATCH --time=2-00:00:00 #Set the wall clock limit to 1hr and 30min
#SBATCH --ntasks=1 #Request 1 task
#SBATCH --cpus-per-task=48 #Request 1 task
#SBATCH --mem=50G #Request 100GB per node
module purge
ml iccifort/2019.5.281 impi/2018.5.288 OrthoFinder/2.3.11-Python-3.7.4
ml IQ-TREE/1.6.12 FastTree/2.1.11
ml MAFFT/7.453-with-extensions
proteome_dir="/scratch/group/songlab/maeva/LocustsGenomeEvolution/Version3/5_OrthoFinder/fasta"
temporary_dir="/scratch/group/songlab/maeva/LocustsGenomeEvolution/tmp"
# Check if directories exist
if [[ ! -d $proteome_dir ]]; then
echo "Proteome directory $proteome_dir does not exist. Exiting."
exit 1
fi
if [[ ! -d $temporary_dir ]]; then
echo "Temporary directory $temporary_dir does not exist. Creating it now."
mkdir -p $temporary_dir
fi
# Run OrthoFinder
orthofinder -S diamond \
-T iqtree \
-A mafft \
-a 24 \
-I 1.5 \
-t 24 \
-M msa \
-f "$proteome_dir" \
-p "$temporary_dir"Here we use:
-S diamond DIAMOND as a sequence search program
-T iqtree IQTREE as a tree inference program
-A mafft MAFFT as the multiple sequence alignment (MSA)
program
-I 1.5 MCL inflation parameter (default from the
pipeline)
-t 32 number of threads
If you expect tightly conserved orthogroups (e.g., highly conserved core genes), consider a higher inflation value (e.g., -I 2.0 or even -I 3.0). This will favor clusters with tighter connections, reducing the possibility of grouping genes that diverge functionally.
If you’re studying functionally diverse or rapidly evolving gene families (e.g., gene families with species-specific expansions), a lower inflation value (e.g., -I 1.2 to -I 1.5) may help retain related genes in the same orthogroup, even if they have evolved to some degree.
We will run this analysis with also the -I 3 since we
expect close relation among Orthoptera genes.
If we run orthofinder using BLAST as it can give -2% accuracy increase over DIAMOND but will be computationally more heavy. For this we will only change:
ml BLAST+/2.9.0
orthofinder -S blastand we can run the script with the blast, iqtree and mafft options:
sbatch scripts/orthofinder_blast.shNow we are all done, we can explore the results and go on for the next steps.
6. Orthogroups to GeneID
At the end we obtain several folders, for which the content is extremely well explain by the software developer David Emms here.
Briefly, one of the important output in the folder
Orthogroups is the actual Orthogroups.txt
file. Although other files there are also important as we can use
Orthogroups.GeneCount.tsv for CAFE5 later on,
for example.
One of the issue with the pipeline we use is that, there will be some
suffix in front of our protein coding, so we will remove that by using
the following python code conversion_ortho.py:
import re
import csv
# === Process TXT file ===
input_file_txt = 'Orthogroups.txt'
output_file_txt = 'Orthogroups_reprocessed.txt'
# Read the input file
with open(input_file_txt, 'r') as file:
data = file.read()
# Remove all prefixes before "_XP", "_NP", and "_YP"
result = re.sub(r'\b\w+_(XP|NP|YP)', r'\1', data)
# Write the result to the output file
with open(output_file_txt, 'w') as file:
file.write(result)
print(f"Processed data has been written to {output_file_txt}")
# === Process TSV file ===
input_file_tsv = 'Orthogroups.tsv'
output_file_tsv = 'Orthogroups_reprocessed.tsv'
# Open the input and output files
with open(input_file_tsv, 'r') as infile, open(output_file_tsv, 'w', newline='') as outfile:
# Create CSV reader and writer for TSV
reader = csv.reader(infile, delimiter='\t')
writer = csv.writer(outfile, delimiter='\t')
# Process each row
for row in reader:
# Modify each field in the row
modified_row = [re.sub(r'\b\w+_(XP|NP|YP)', r'\1', field) for field in row]
# Write the modified row to the output
writer.writerow(modified_row)
print(f"Processed data has been written to {output_file_tsv}")Note: Inspect the reprocessed file. I found that some protein_id have changed over time, for example some protein in nitens got an appended “_p1/7”, and some the proteins ID may have a “_1/_2” in one file and “.1/.2” in other files. Make sure to check your table after each merging.
We used the python script python protein2geneid_loop.py
written by David to extract the protein_id from each gene_id and make a
full table with all species. For this we need to run the script in the
folder 1_RawData.
import os
import re
# Get the current working directory
gff_directory = os.getcwd()
# Create a directory to store the output files
output_directory = os.path.join(gff_directory, "output_files")
os.makedirs(output_directory, exist_ok=True)
# List GFF files with "_GFF.gff" extension
species_list = [filename for filename in os.listdir(gff_directory) if filename.endswith("_GFF.gff")]
# Process each species' GFF file
for species_filename in species_list:
# Extract the species name from the file name
species_name = re.sub(r"_GFF\.gff$", "", species_filename)
# Construct input and output file paths
input_path = os.path.join(gff_directory, species_filename)
output_filename = f"xp{species_name}.gff" # Remove the dot before species_name
output_path = os.path.join(output_directory, output_filename)
# Use 'grep' to filter lines containing "XP" and save to the output file
grep_command = f'grep "XP" "{input_path}" > "{output_path}"'
os.system(grep_command)
# Print a message indicating the filtering process
print(f"Filtered {species_filename} to {output_filename}")
# Construct the output TSV file path
tsv_output_filename = f'gffKey{species_name}.tsv' # Remove the dot before species_name
tsv_output_path = os.path.join(output_directory, tsv_output_filename)
lol = {}
# Read and process the contents of the GFF file
with open(output_path) as gffFile:
for line in gffFile:
if re.search(r"product=[^;]+", line):
gene_match = re.search(r"gene=[^;=]+", line)
product_match = re.search(r"product=[^;]+", line)
proteinID_match = re.search(r"protein_id=(.+)", line)
if gene_match and product_match and proteinID_match:
gene = gene_match.group(0)
if gene.endswith('product'):
gene = gene[5:-7]
else:
gene = gene[5:]
product = product_match.group(0)
product = product.replace("product=", "")
product = product.rstrip()
proteinID = proteinID_match.group(1)
if proteinID not in lol.keys():
lol[proteinID] = [gene, product, species_name] # Add species name
# Write the processed data to the output TSV file
with open(tsv_output_path, 'w') as output:
for proID, lis in lol.items():
output.writelines(proID + '\t' + lis[0] + '\t' + lis[1] + '\t' + lis[2] + '\n') # Include species name
# Print a message indicating the processing of the GFF file
print(f"Processed {output_filename} to {tsv_output_filename}")
# Concatenate all final files into a single file
final_output_filename = "allspecies_protein2geneid.tsv"
final_output_path = os.path.join(output_directory, final_output_filename)
with open(final_output_path, 'w') as final_output:
for species_filename in species_list:
species_name = re.sub(r"_GFF\.gff$", "", species_filename)
tsv_output_filename = f'gffKey{species_name}.tsv' # Remove the dot before species_name
tsv_output_path = os.path.join(output_directory, tsv_output_filename)
with open(tsv_output_path, 'r') as species_output:
final_output.write(species_output.read())
print(f"Concatenated all species files into {final_output_filename}")
Launch the script as python protein2geneid_loop.py.
Once we have obtained both files, we can join them with R and we will have a correspondence among orthogroups_id, protein_id, gene_id, gene description and species. I had to sort the processed file by hand with excel for the first 6 Orthogroups (too long line, cutting names).
NB: I noticed there are still some small issues with the file
allspecies_protein2geneid.tsv so I fixed it by hand
afterwards. NB2: We can either filter for Schistocerca only or
we can also just run it on the Schistocerca only Orthofinder
output (difference is -I 2 vs -I 5)
7. Orthofinder Results
Schistocerca only

library(cogeqc)
library(ggtree)
library(treeio)
library(dplyr)
library(ggplot2)
library(stringr)
# Set the base directory for your Orthofinder results
ortho_dir <- "/Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Schistocerca/Results_I2/"
# Load the orthogroup file
orthogroups <- read_orthogroups(file.path(ortho_dir, "Orthogroups/Orthogroups_reprocessed.tsv"))
# Remove "_filteredTranscripts" from the Species column
orthogroups <- orthogroups %>%
mutate(SpeciesID = str_replace(Species, "_filteredTranscripts", "")) %>% # Clean Species name
select(Orthogroup, SpeciesID, Gene) # Rename and keep relevant columns
# Check the first few rows
head(orthogroups) Orthogroup SpeciesID Gene
1 OG0000000 Samer XP_046979578.1
2 OG0000000 Samer XP_046980609.1
3 OG0000000 Samer XP_046980698.1
4 OG0000000 Samer XP_046981444.1
5 OG0000000 Samer XP_046981490.1
6 OG0000000 Samer XP_046982927.1# Load the directory with the actual stats from the Orthofinder run
ortho_stats <- read_orthofinder_stats(file.path(ortho_dir, "Comparative_Genomics_Statistics"))
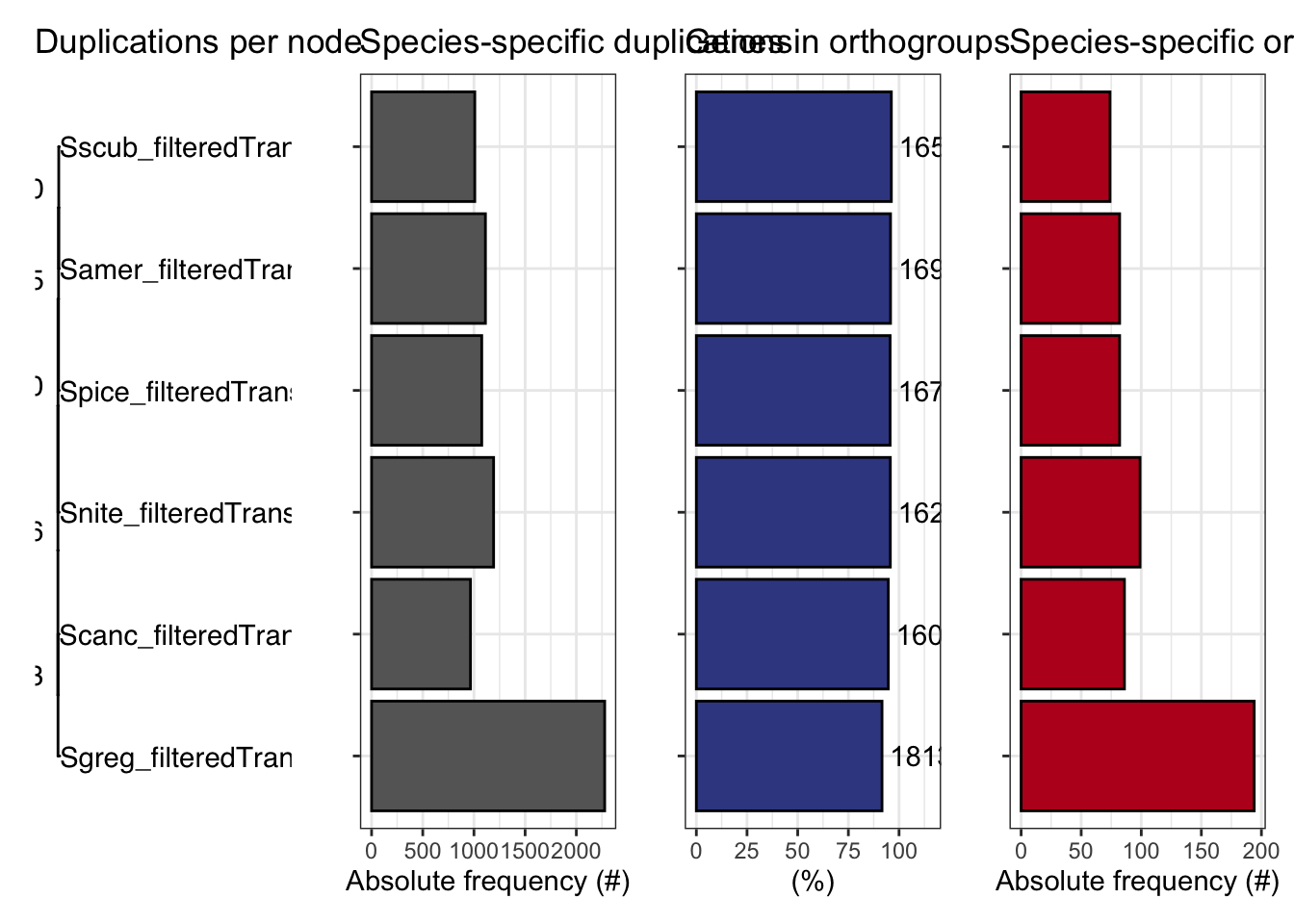
ortho_stats$stats Species N_genes N_genes_in_OGs Perc_genes_in_OGs N_ssOGs
1 Samer_filteredTranscripts 17661 16918 95.8 82
2 Scanc_filteredTranscripts 16906 16008 94.7 86
3 Sgreg_filteredTranscripts 19798 18139 91.6 194
4 Snite_filteredTranscripts 16935 16215 95.7 99
5 Spice_filteredTranscripts 17489 16722 95.6 82
6 Sscub_filteredTranscripts 17236 16588 96.2 74
N_genes_in_ssOGs Perc_genes_in_ssOGs Dups
1 515 2.9 1112
2 364 2.2 966
3 721 3.6 2277
4 612 3.6 1192
5 393 2.2 1076
6 435 2.5 1008tree <- treeio::read.tree(file.path(ortho_dir, "Species_Tree/SpeciesTree_rooted_node_labels.txt"))
#tree$tip.label
#custom plot_species
plot_species_tree <- function(tree = NULL, xlim = c(0, 2), stats_list = NULL, custom_labels = NULL) {
# Basic tree plot with customized theme
p <- ggtree(tree) +
xlim(xlim) +
theme_tree() + # Use a clean theme
ggtitle("Species Tree with Duplications") + # Set a title
theme(plot.title = element_text(hjust = 0.5)) # Center the title
# Customize tip labels if provided
if (!is.null(custom_labels)) {
# Ensure length of custom_labels matches the number of tree tips
if (length(custom_labels) == length(tree$tip.label)) {
p <- p + geom_tiplab(aes(label = custom_labels), size = 4, fontface = "bold.italic", color = "darkblue")
} else {
stop("Length of custom_labels must match the number of tip labels in the tree.")
}
} else {
# Default tip labels if no custom labels are provided
p <- p + geom_tiplab(size = 4, fontface = "bold.italic", color = "darkblue")
}
if (!is.null(stats_list)) {
# Extract duplications
dups <- stats_list$duplications
dups <- dups[dups$Node %in% tree$tip.label, ] # Filter for relevant nodes
names(dups) <- c("label", "dups")
# Check for matching nodes
if (nrow(dups) > 0) {
p$data <- merge(p$data, dups, by.x = "label", by.y = "label", all.x = TRUE)
# Add duplications to the plot with larger text
p <- p +
ggtree::geom_text2(
aes(label = .data$dups),
hjust = 1.3, vjust = -0.5,
size = 5, color = "red" # Customize size and color of duplication labels
) +
labs(subtitle = "Number of Duplications per Node") # Subtitle for clarity
} else {
message("No matching nodes found for duplications.")
}
}
# Add circles around the nodes
p <- p + geom_point(size = 2, shape = 21, color = "black", fill = "black") # Circle around nodes
return(p)
}
# Plotting the tree
labels6 <- c("Schistocerca gregaria", "Schistocerca piceifrons", "Schistocerca americana", "Schistocerca serialis cubense", "Schistocerca cancellata", "Schistocerca nitens")
# Call the custom plot function with your species tree, stats, and custom labels
p<- plot_species_tree(tree, xlim = c(0, 1.5), stats_list = ortho_stats)
plot_duplications(ortho_stats)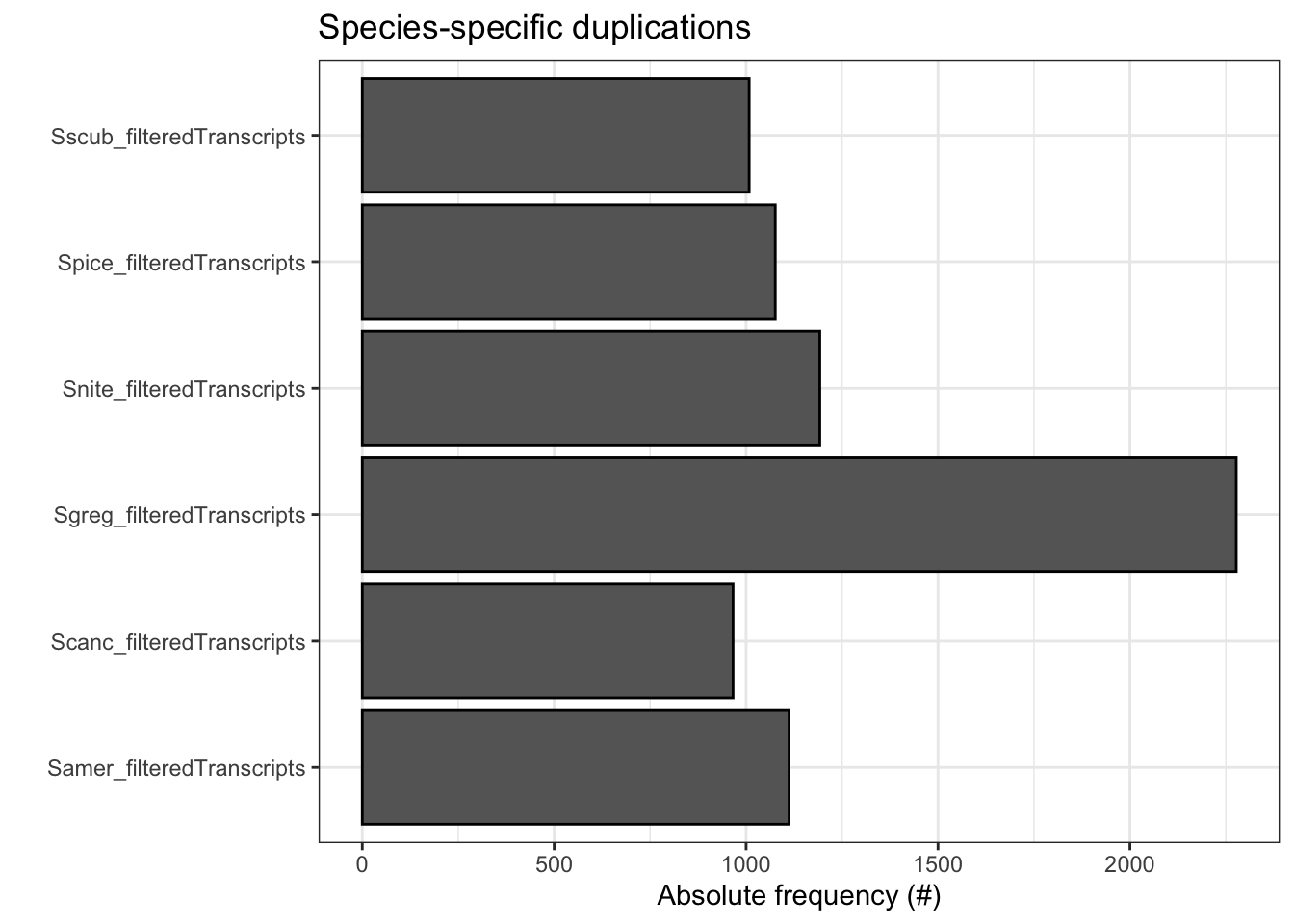
plot_genes_in_ogs(ortho_stats)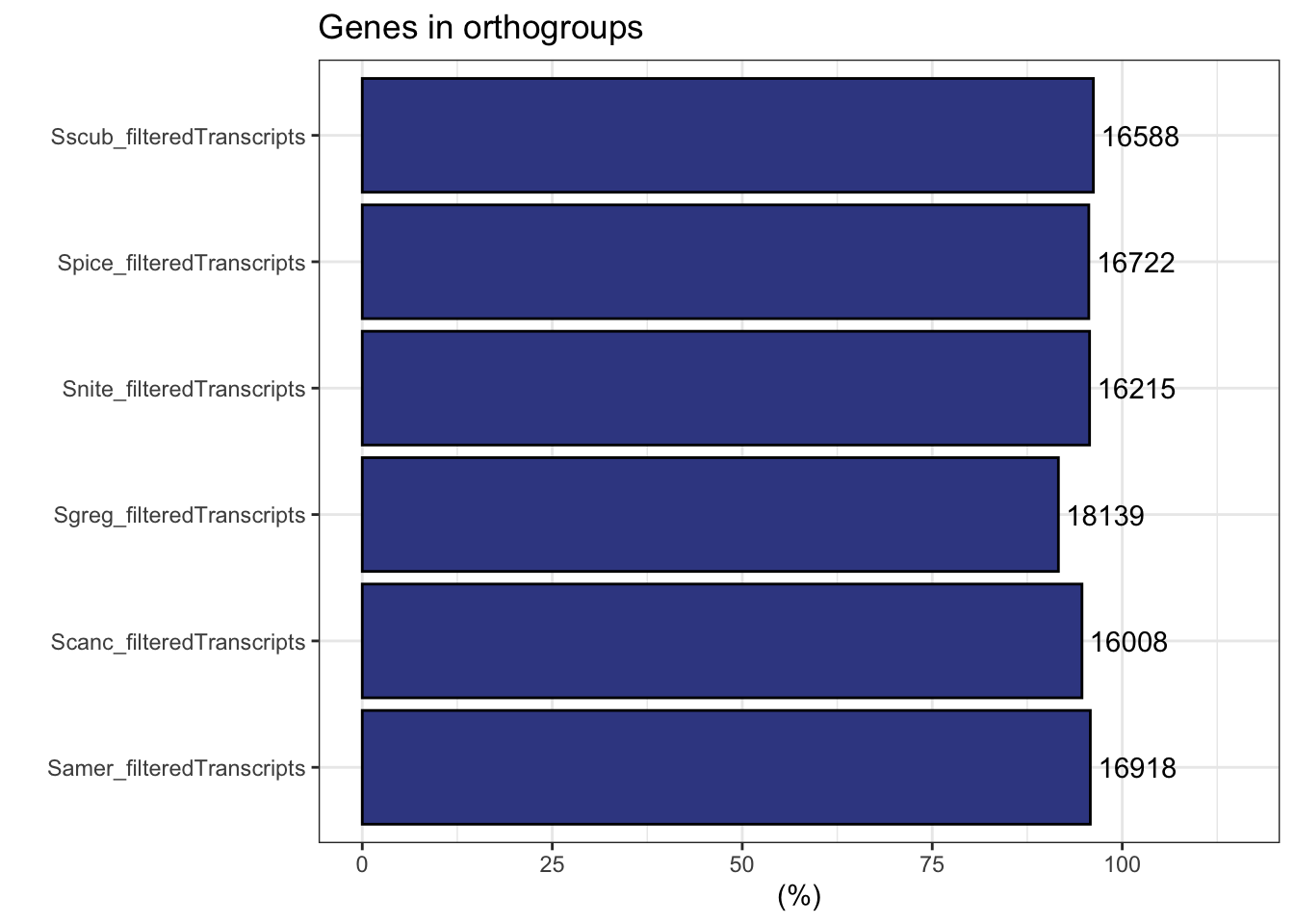
plot_species_specific_ogs(ortho_stats)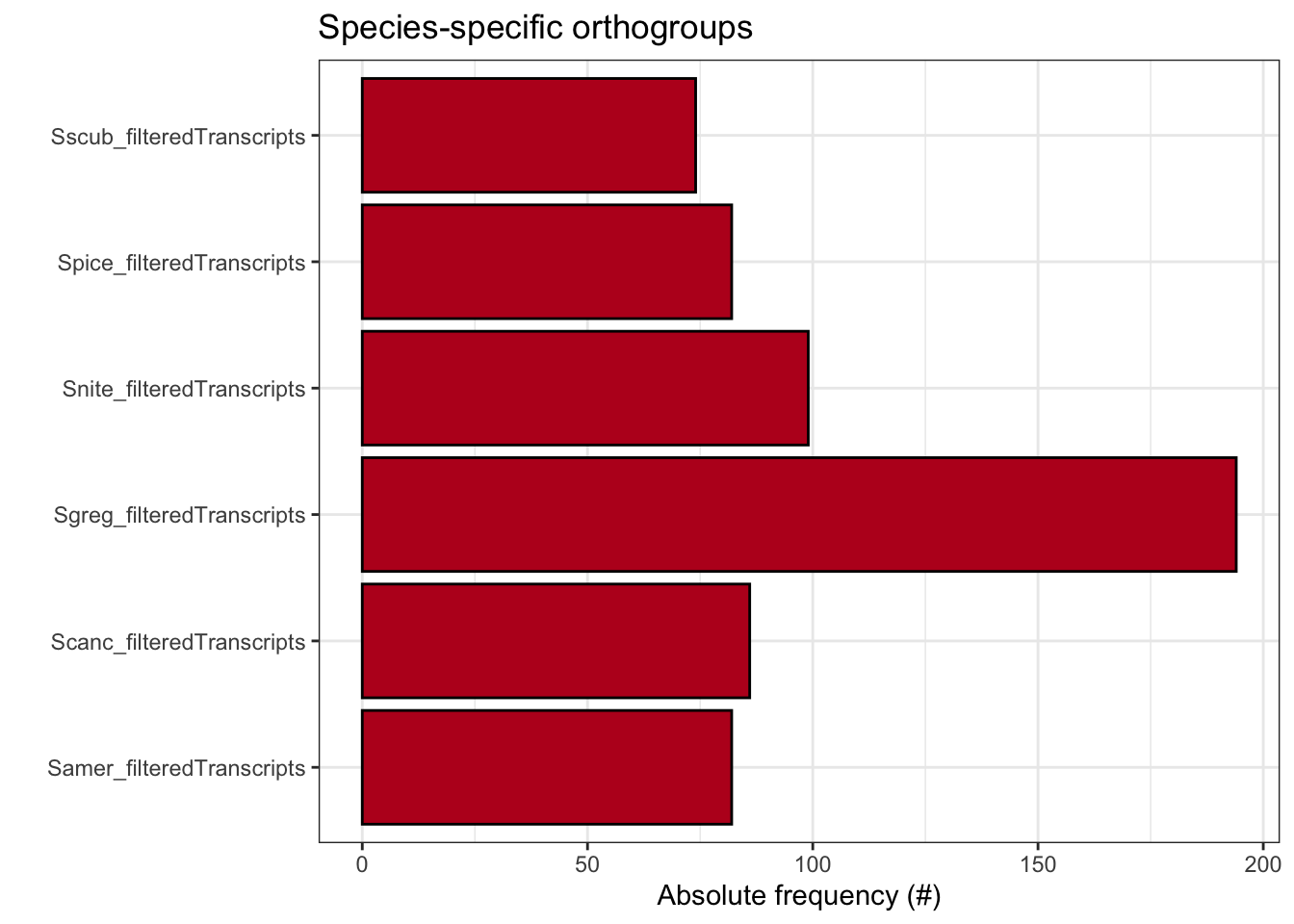
plot_orthofinder_stats(
tree = tree,
xlim = c(-0.1, 2),
stats_list = ortho_stats
)
plot_og_overlap(ortho_stats)
plot_og_sizes(orthogroups)
plot_og_sizes(orthogroups, log = TRUE) 
library(ggplot2)
library(dplyr)
library(tidyr)
library(stringr)
library(tibble)
# Set the base directory for your Orthofinder results
ortho_dir <- "/Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Schistocerca/Results_I2/"
dResults <- paste0(ortho_dir, "Comparative_Genomics_Statistics/")
files <- c(
"OrthologuesStats_one-to-one.tsv",
"OrthologuesStats_one-to-many.tsv",
"OrthologuesStats_many-to-one.tsv",
"OrthologuesStats_many-to-many.tsv"
)
# === Function to Read and Process Data ===
read_data_matrix <- function(file_path) {
data <- read.delim(file_path, check.names = FALSE, row.names = 1)
return(as.matrix(data))
}
# === Load Data from Files ===
d11 <- read_data_matrix(paste0(dResults, files[1]))
d1m <- read_data_matrix(paste0(dResults, files[2]))
dm1 <- read_data_matrix(paste0(dResults, files[3]))
dmm <- read_data_matrix(paste0(dResults, files[4]))
# === Schistocerca Species Selection and Order ===
species <- c(
"Sscub_filteredTranscripts", # cubense
"Samer_filteredTranscripts", # americana
"Spice_filteredTranscripts", # piceifrons
"Scanc_filteredTranscripts", # cancellata
"Snite_filteredTranscripts", # nitens
"Sgreg_filteredTranscripts" # gregaria
)
# === Map Species to Readable Labels ===
species_labels <- c(
"Sscub_filteredTranscripts" = "cubense",
"Samer_filteredTranscripts" = "americana",
"Spice_filteredTranscripts" = "piceifrons",
"Scanc_filteredTranscripts" = "cancellata",
"Snite_filteredTranscripts" = "nitens",
"Sgreg_filteredTranscripts" = "gregaria"
)
# === Update Row Names ===
rownames(d11) <- species
rownames(d1m) <- species
rownames(dm1) <- species
rownames(dmm) <- species
# === Function to Prepare Data (Self-Comparison Blank) ===
combine_data <- function(d11, d1m, dm1, dmm, species, species_to_plot) {
isp <- which(species == species_to_plot)
dtot <- d11 + d1m + dm1 + dmm
data <- data.frame(
Species = species,
`1:1` = ifelse(species == species_to_plot, 0, d11[isp, ] / dtot[isp, ] * 100),
`1:many` = ifelse(species == species_to_plot, 0, d1m[isp, ] / dtot[isp, ] * 100),
`many:1` = ifelse(species == species_to_plot, 0, dm1[isp, ] / dtot[isp, ] * 100),
`many:many` = ifelse(species == species_to_plot, 0, dmm[isp, ] / dtot[isp, ] * 100)
)
return(data)
}
# === Function to Create and Save Stacked Bar Plot (Vertical) ===
create_plot <- function(data, species_to_plot, output_dir = ortho_dir) {
data_long <- data %>%
pivot_longer(
cols = -Species,
names_to = "Relationship",
values_to = "Proportion"
)
# Rename relationships to match expected levels
data_long$Relationship <- str_replace_all(data_long$Relationship, c(
"X1.1" = "1:1",
"X1.many" = "1:many",
"many.1" = "many:1",
"many.many" = "many:many"
))
# Ensure Relationship column is a factor with consistent levels
data_long$Relationship <- factor(
data_long$Relationship,
levels = c("1:1", "1:many", "many:1", "many:many")
)
# Set Custom Species Order
data_long$Species <- factor(data_long$Species, levels = species, labels = species_labels)
# Generate the Plot
plot <- ggplot(data_long, aes(x = Species, y = Proportion, fill = Relationship)) +
geom_bar(stat = "identity", position = "stack") +
scale_fill_manual(
values = c(
`1:1` = "forestgreen",
`1:many` = "orange",
`many:1` = "purple",
`many:many` = "black"
)
) +
labs(
title = paste("Orthologue multiplicty vs", species_labels[species_to_plot]),
x = "Species",
y = "Proportion (%)",
fill = "Relationship Type"
) +
theme_minimal() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1),
axis.title.x = element_text(size = 12),
axis.title.y = element_text(size = 12)
)+
coord_flip()
# Save the Plot
output_file <- paste0(output_dir, "VerticalStackedBar_", species_labels[species_to_plot], ".pdf")
ggsave(output_file, plot = plot, width = 10, height = 6)
cat("Plot saved as:", output_file, "\n")
}
# === Generate Vertical Plots for Each Schistocerca Species ===
output_dir <- paste0(ortho_dir, "Plots_Schistocerca/") # Specify output directory
dir.create(output_dir, showWarnings = FALSE)
for (species_to_plot in species) {
data <- combine_data(d11, d1m, dm1, dmm, species, species_to_plot)
create_plot(data, species_to_plot, output_dir)
}Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Schistocerca/Results_I2/Plots_Schistocerca/VerticalStackedBar_cubense.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Schistocerca/Results_I2/Plots_Schistocerca/VerticalStackedBar_americana.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Schistocerca/Results_I2/Plots_Schistocerca/VerticalStackedBar_piceifrons.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Schistocerca/Results_I2/Plots_Schistocerca/VerticalStackedBar_cancellata.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Schistocerca/Results_I2/Plots_Schistocerca/VerticalStackedBar_nitens.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Schistocerca/Results_I2/Plots_Schistocerca/VerticalStackedBar_gregaria.pdf library(readr)
# rename for future merging
names(orthogroups)[names(orthogroups) == "Gene"] <- "protein_id"
orthogroups <- orthogroups %>%
mutate(protein_id = str_remove(protein_id, "_p[1-7]$"))
# Optional if you did not rename fasta sequence before
#orthogroups$Species <- gsub("_filteredproteome", "", orthogroups$SpeciesID)
# Export the table to tab-separated text file (you can change the delimiter if needed)
output_file <- file.path(ortho_dir, "Orthogroups_Schistocerca_Jan2025.txt")
# Write the transformed data to the file
write.table(orthogroups,
file = output_file,
sep = "\t",
quote = FALSE,
row.names = FALSE)
proteingene_path <- file.path(ortho_dir, "../../../list/allspecies_protein2geneid.tsv")
proteingeneid <- read_tsv(proteingene_path, col_names = TRUE )
head(proteingeneid)# A tibble: 6 × 4
protein_id GeneID Description Species
<chr> <chr> <chr> <chr>
1 XP_047114676.1 LOC124794980 piggyBac transposable element-derived pro… Schist…
2 XP_047114773.1 LOC124795035 piggyBac transposable element-derived pro… Schist…
3 XP_047117829.1 LOC124798450 uncharacterized protein LOC124798450 Schist…
4 XP_047118606.1 LOC124799109 RNA-binding protein 25-like Schist…
5 XP_047118853.1 LOC124799306 molybdenum cofactor biosynthesis protein … Schist…
6 XP_047118904.1 LOC124799809 molybdenum cofactor biosynthesis protein … Schist…biotype_path <- file.path(ortho_dir, "../../../list/13polyneoptera_geneid_ncbi.csv")
biotypeid <- read_csv(biotype_path, col_names = TRUE )
head(biotypeid)# A tibble: 6 × 10
Accession Begin End Description Symbol GeneID GeneType Transcripts_accession
<chr> <dbl> <dbl> <chr> <chr> <chr> <chr> <chr>
1 NC_06468… 1 66 tRNA-Ile trnI LOC73… tRNA <NA>
2 NC_06468… 70 138 tRNA-Gln trnQ LOC73… tRNA <NA>
3 NC_06468… 138 206 tRNA-Met trnM LOC73… tRNA <NA>
4 NC_06468… 207 1227 NADH dehyd… ND2 LOC73… protein… <NA>
5 NC_06468… 1228 1295 tRNA-Trp trnW LOC73… tRNA <NA>
6 NC_06468… 1288 1350 tRNA-Cys trnC LOC73… tRNA <NA>
# ℹ 2 more variables: protein_id <chr>, Species <chr># Ensure GeneID is treated as a character to avoid mismatches
proteinorthotable <- left_join(orthogroups, proteingeneid, by = "protein_id")
final_orthotable <- left_join(biotypeid, proteinorthotable[, c("Orthogroup","SpeciesID", "GeneID")], by = "GeneID")
output_file <- file.path(ortho_dir, "Orthogroups_genesproteinbiotype_Schistocerca_Jan2025.csv")
# Write the table with proper quoting and no row names
write.table(final_orthotable, file = output_file, sep = ",", quote = TRUE, row.names = FALSE, col.names = TRUE)There we have it, the final table with all the corresponding IDs. This table is too big and have all species in it, so we want to reduce to only single copy orthologs and remove entries with other species than Schistocerca for now.
# Filter the final_orthotable to keep only rows with species 'Schistocerca'
filtered_final_orthotable <- final_orthotable %>%
filter(Species %in% c("Schistocerca gregaria", "Schistocerca piceifrons", "Schistocerca americana", "Schistocerca cancellata", "Schistocerca serialis cubense", "Schistocerca nitens"))
# Optionally, save the filtered data
#output_file <- file.path(ortho_dir, "Orthogroups_genesprotein_Schisto_Jan2025.txt")
#write.table(filtered_final_orthotable, file = output_file, sep = "\t", quote = FALSE, row.names = FALSE)
# Step 1: Read the single copy orthologs table
single_copy_orthologs_path <- file.path(ortho_dir, "Orthogroups/Orthogroups_SingleCopyOrthologues.txt")
single_copy_orthologs <- read.table(single_copy_orthologs_path, header = FALSE, stringsAsFactors = FALSE)
# Step 2: Ensure the column name for orthogroups matches in both data frames
# If necessary, rename the column in single_copy_orthologs to match
colnames(single_copy_orthologs) <- c("Orthogroup") # Replace with the actual name if different
# Step 3: Perform the intersection
scopy_final_orthotable <- final_orthotable[final_orthotable$Orthogroup %in% single_copy_orthologs$Orthogroup, ]
# Step 4: Optionally, save the filtered table
output_file <- file.path(ortho_dir, "SingleCopyOrthogroups_genesprotein_6species_Jan2025.txt")
write.table(scopy_final_orthotable, file = output_file, sep = "\t", quote = FALSE, row.names = FALSE)Polyneoptera
library(cogeqc)
library(ggtree)
library(treeio)
library(dplyr)
library(ggplot2)
# Set the base directory for your Orthofinder results
ortho_dir <- "/Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/"
# Load the orthogroup file
orthogroups <- read_orthogroups(file.path(ortho_dir, "Orthogroups/Orthogroups_reprocessed.tsv"))
# Remove "_filteredTranscripts" from the Species column
orthogroups <- orthogroups %>%
mutate(SpeciesID = str_replace(Species, "_filteredTranscripts", "")) %>% # Clean Species name
select(Orthogroup, SpeciesID, Gene) # Rename and keep relevant columns
# Load the directory with the actual stats from the Orthofinder run
ortho_stats <- read_orthofinder_stats(file.path(ortho_dir, "Comparative_Genomics_Statistics"))
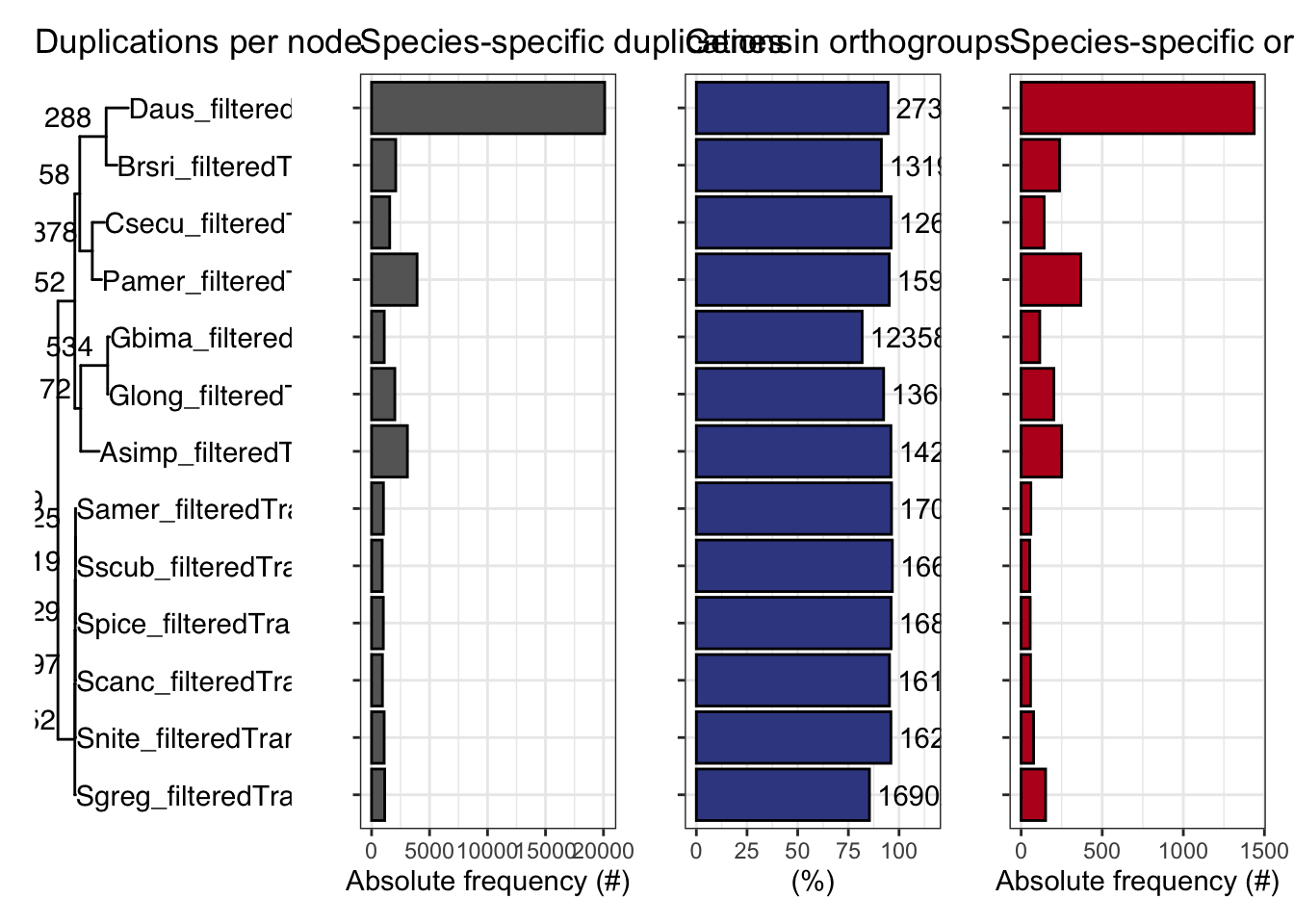
ortho_stats$stats Species N_genes N_genes_in_OGs Perc_genes_in_OGs N_ssOGs
1 Asimp_filteredTranscripts 14865 14272 96.0 250
2 Brsri_filteredTranscripts 14447 13195 91.3 237
3 Csecu_filteredTranscripts 13169 12653 96.1 142
4 Daus_filteredTranscripts 28958 27396 94.6 1437
5 Gbima_filteredTranscripts 15112 12358 81.8 115
6 Glong_filteredTranscripts 14729 13604 92.4 202
7 Pamer_filteredTranscripts 16749 15946 95.2 368
8 Samer_filteredTranscripts 17661 17008 96.3 60
9 Scanc_filteredTranscripts 16906 16106 95.3 58
10 Sgreg_filteredTranscripts 19798 16902 85.4 151
11 Snite_filteredTranscripts 16935 16262 96.0 77
12 Spice_filteredTranscripts 17489 16809 96.1 56
13 Sscub_filteredTranscripts 17236 16661 96.7 53
N_genes_in_ssOGs Perc_genes_in_ssOGs Dups
1 1565 10.5 3089
2 1018 7.0 2083
3 679 5.2 1562
4 13979 48.3 20104
5 335 2.2 1092
6 1010 6.9 2006
7 2441 14.6 3924
8 338 1.9 1022
9 280 1.7 935
10 529 2.7 1131
11 501 3.0 1085
12 281 1.6 1012
13 280 1.6 918tree <- treeio::read.tree(file.path(ortho_dir, "Species_Tree/SpeciesTree_rooted_node_labels.txt"))
#tree$tip.label
#custom plot_species
plot_species_tree <- function(tree = NULL, xlim = c(0, 2), stats_list = NULL, custom_labels = NULL) {
# Basic tree plot with customized theme
p <- ggtree(tree) +
xlim(xlim) +
theme_tree() + # Use a clean theme
ggtitle("Species Tree with Duplications") + # Set a title
theme(plot.title = element_text(hjust = 0.5)) # Center the title
# Customize tip labels if provided
if (!is.null(custom_labels)) {
# Ensure length of custom_labels matches the number of tree tips
if (length(custom_labels) == length(tree$tip.label)) {
p <- p + geom_tiplab(aes(label = custom_labels), size = 4, fontface = "bold.italic", color = "darkblue")
} else {
stop("Length of custom_labels must match the number of tip labels in the tree.")
}
} else {
# Default tip labels if no custom labels are provided
p <- p + geom_tiplab(size = 4, fontface = "bold.italic", color = "darkblue")
}
if (!is.null(stats_list)) {
# Extract duplications
dups <- stats_list$duplications
dups <- dups[dups$Node %in% tree$tip.label, ] # Filter for relevant nodes
names(dups) <- c("label", "dups")
# Check for matching nodes
if (nrow(dups) > 0) {
p$data <- merge(p$data, dups, by.x = "label", by.y = "label", all.x = TRUE)
# Add duplications to the plot with larger text
p <- p +
ggtree::geom_text2(
aes(label = .data$dups),
hjust = 1.3, vjust = -0.5,
size = 5, color = "red" # Customize size and color of duplication labels
) +
labs(subtitle = "Number of Duplications per Node") # Subtitle for clarity
} else {
message("No matching nodes found for duplications.")
}
}
# Add circles around the nodes
p <- p + geom_point(size = 2, shape = 21, color = "black", fill = "black") # Circle around nodes
return(p)
}
# Call the custom plot function with your species tree, stats, and custom labels
p<- plot_species_tree(tree, xlim = c(0, 1.5), stats_list = ortho_stats)
plot_duplications(ortho_stats)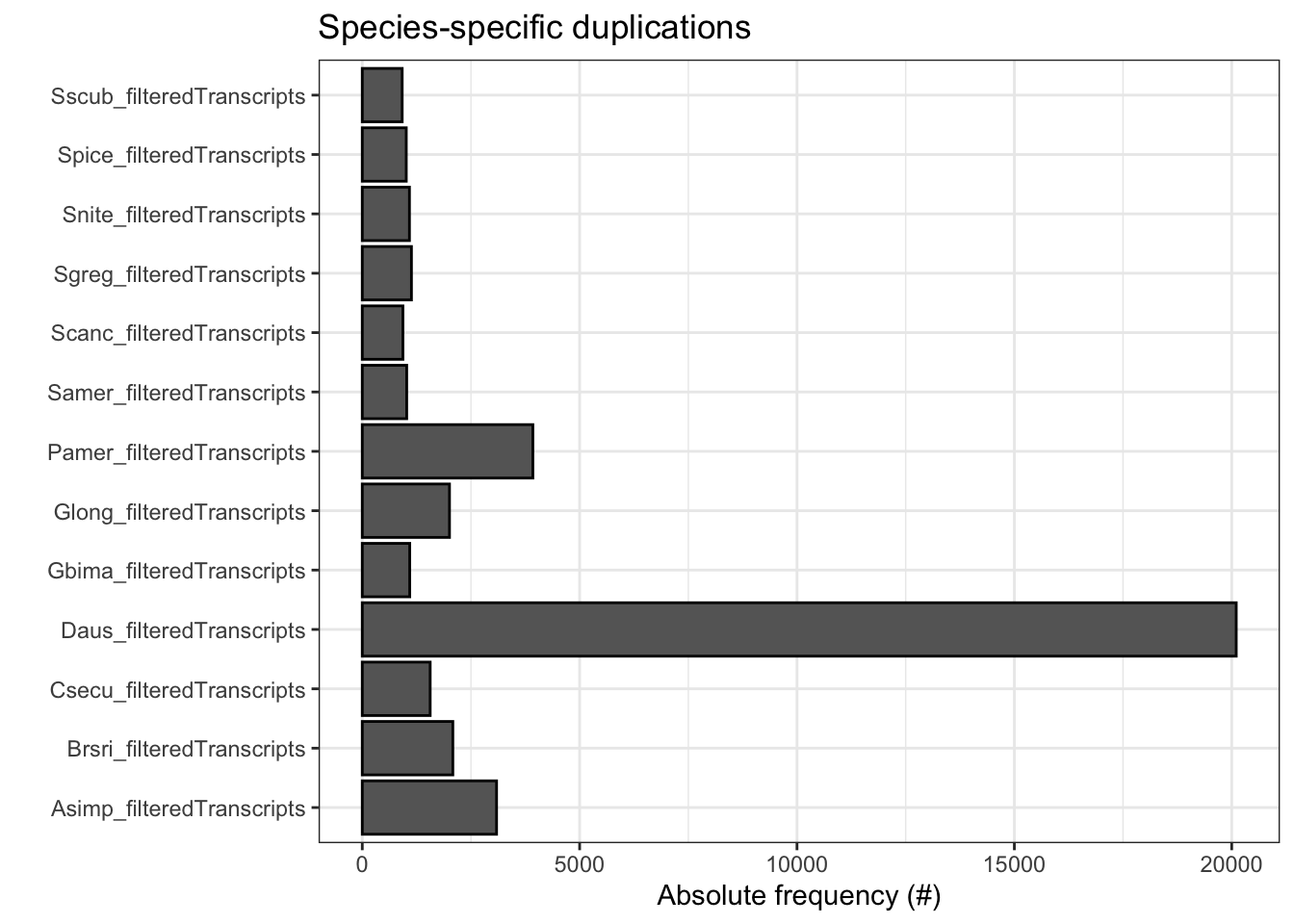
plot_genes_in_ogs(ortho_stats)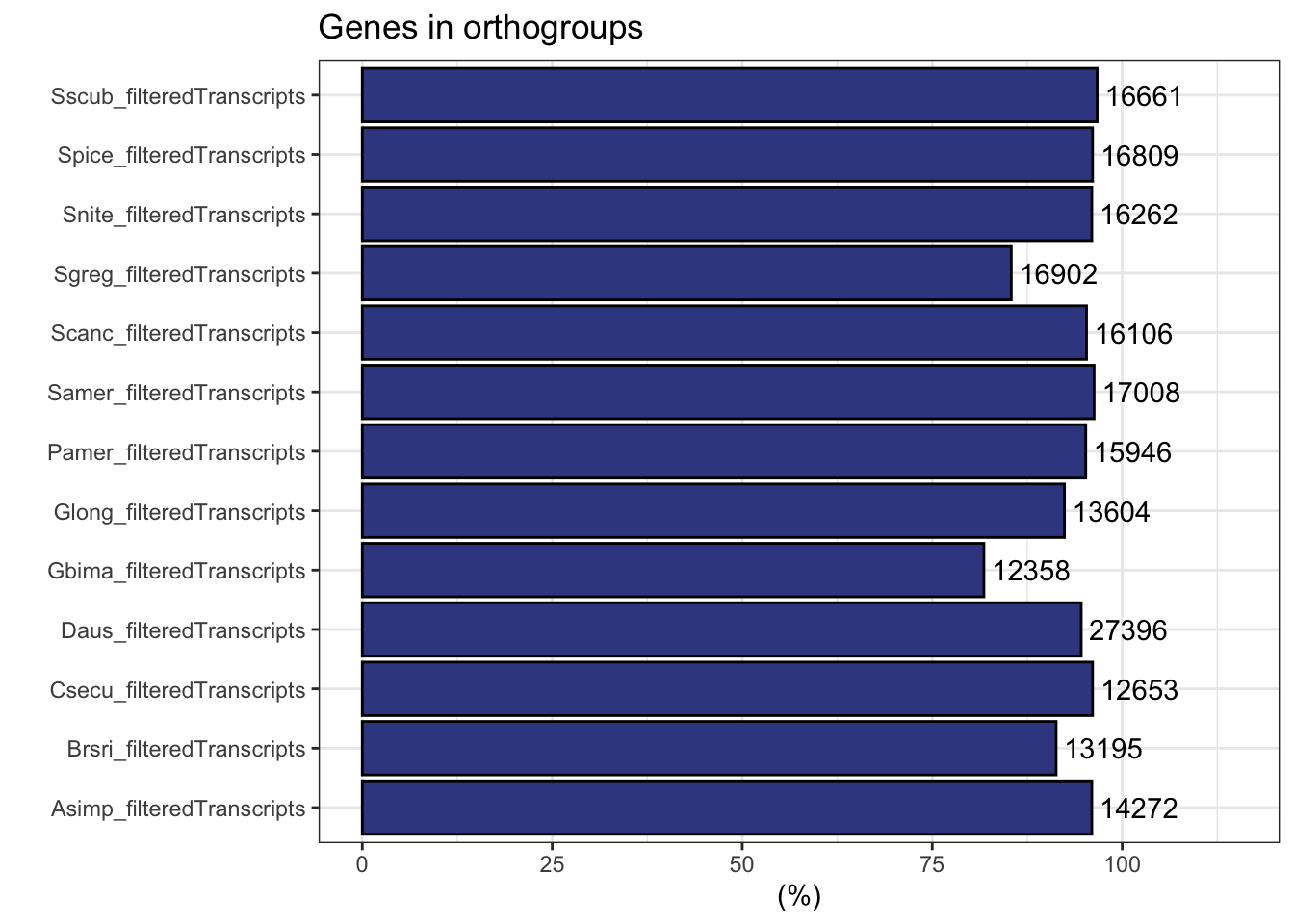
plot_species_specific_ogs(ortho_stats)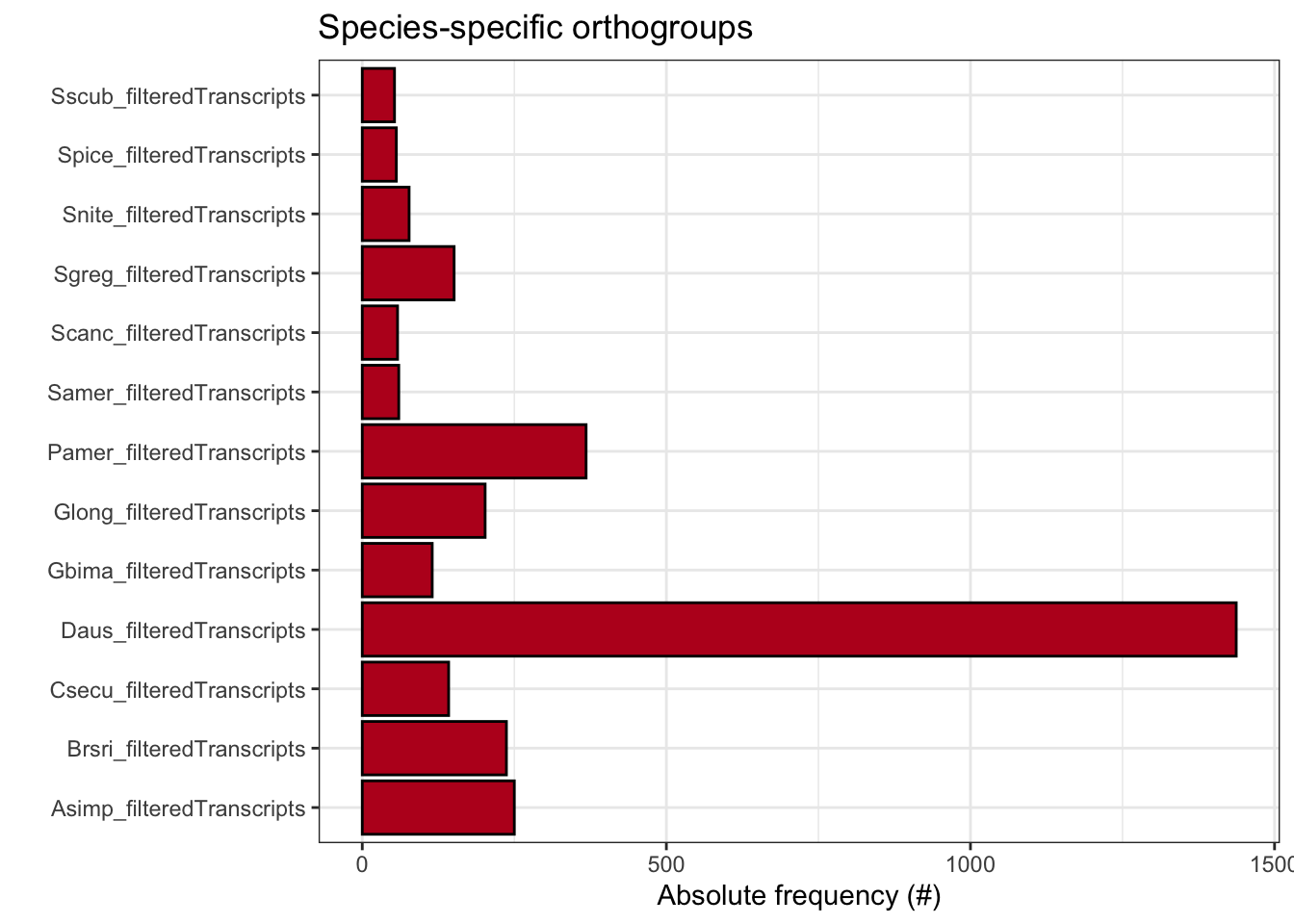
plot_orthofinder_stats(
tree = tree,
xlim = c(-0.1, 2),
stats_list = ortho_stats
)
plot_og_overlap(ortho_stats)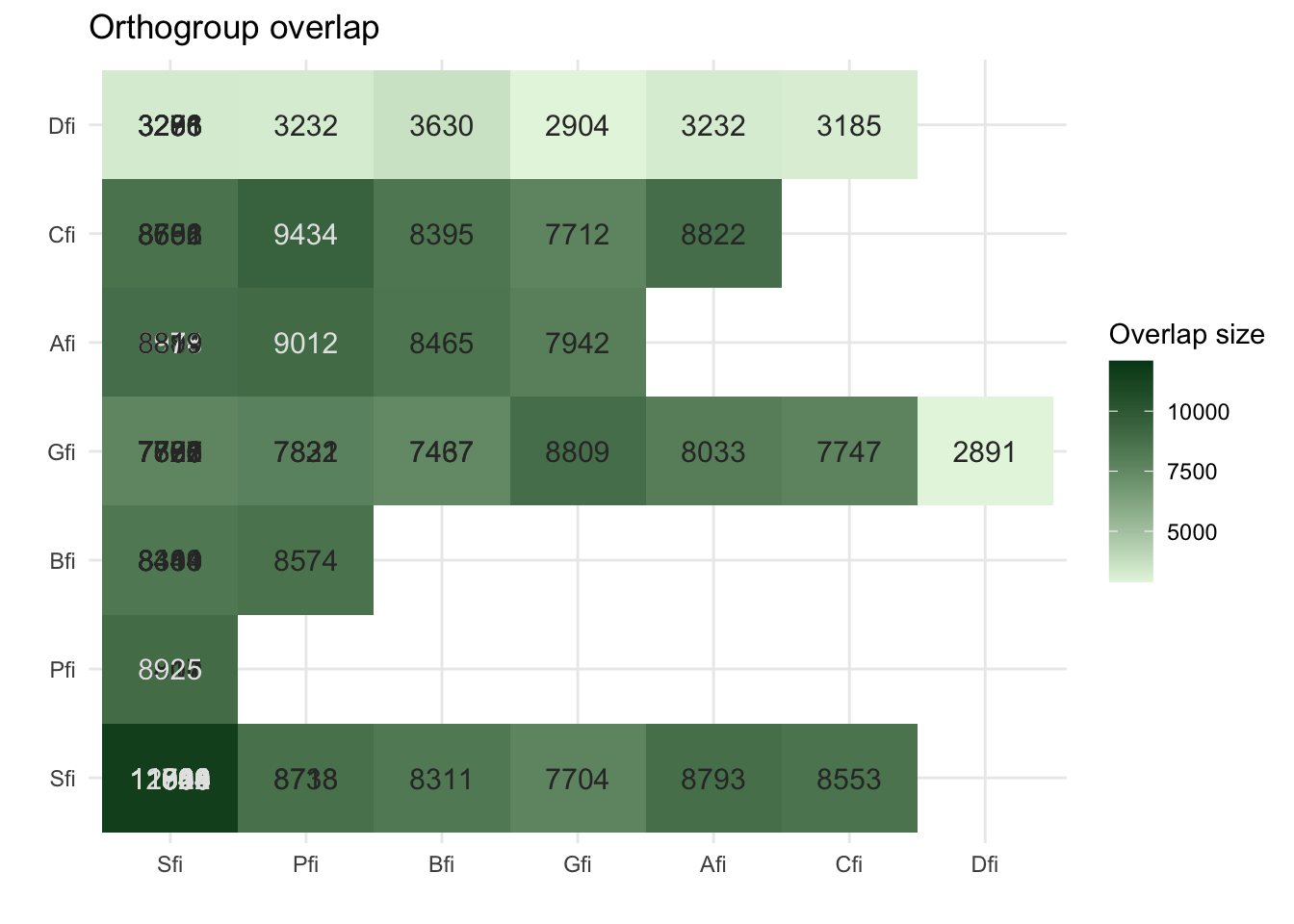
plot_og_sizes(orthogroups)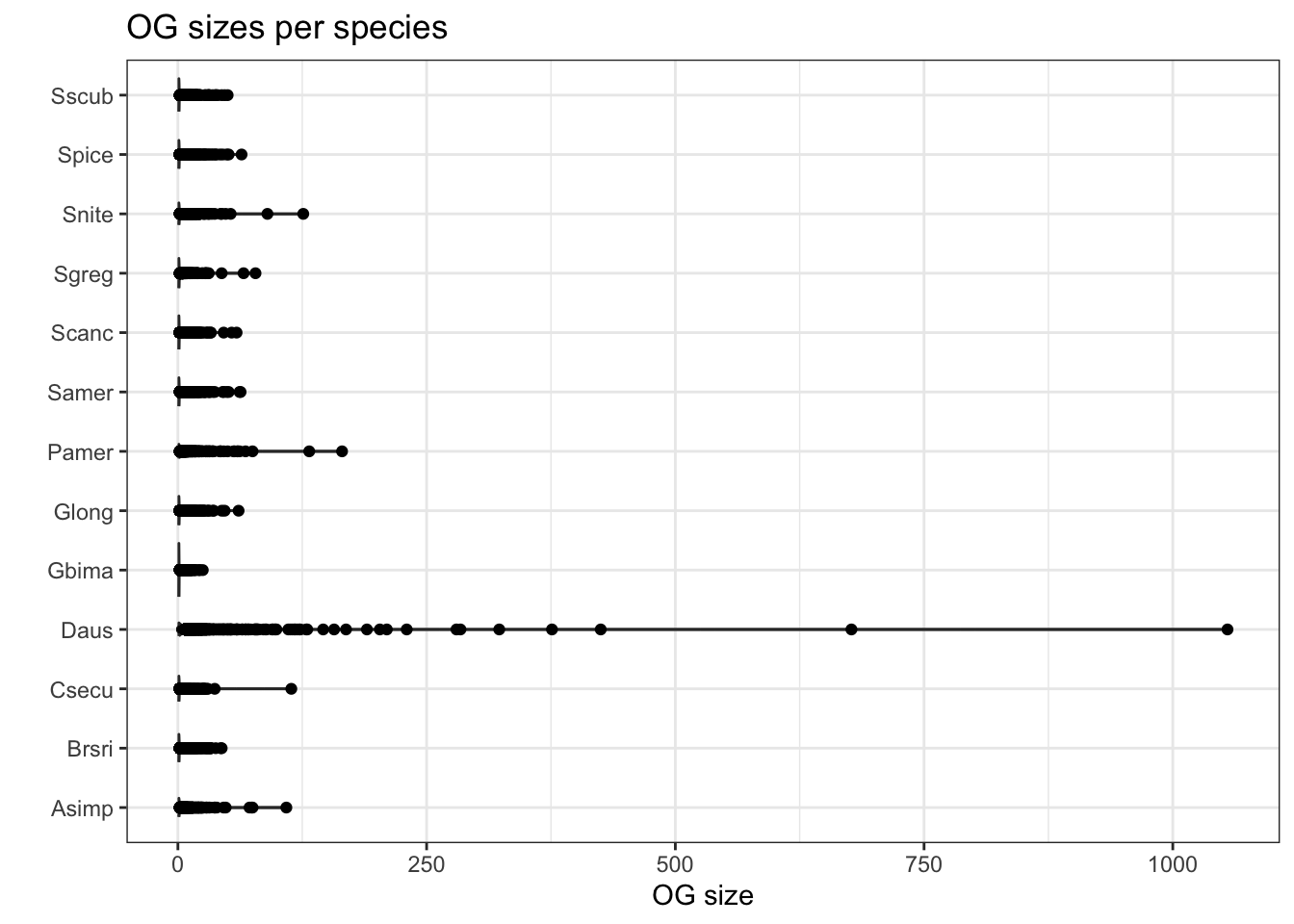
plot_og_sizes(orthogroups, log = TRUE) 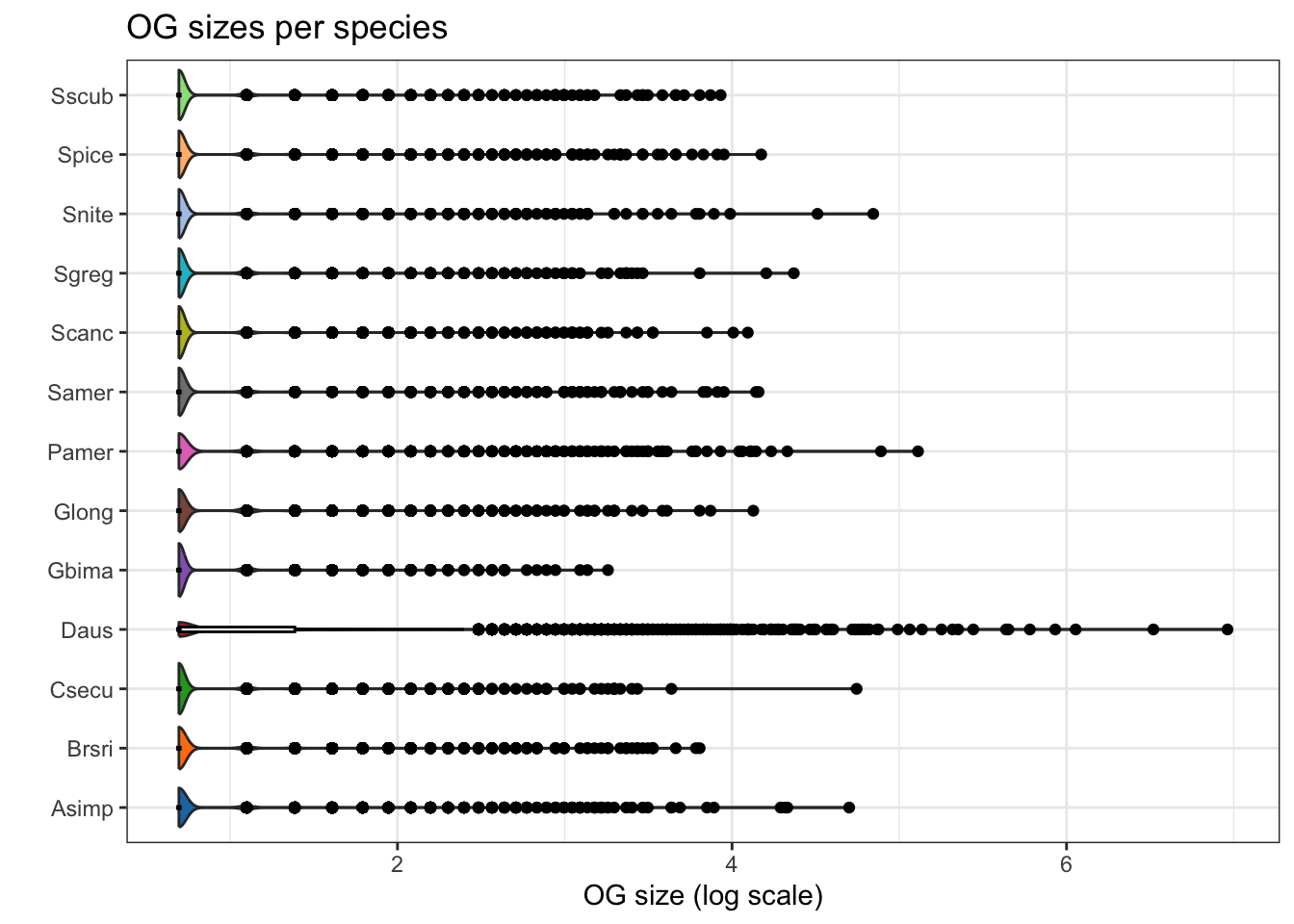
# Set the base directory for your Orthofinder results
ortho_dir <- "/Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/"
dResults <- paste0(ortho_dir, "Comparative_Genomics_Statistics/")
files <- c(
"OrthologuesStats_one-to-one.tsv",
"OrthologuesStats_one-to-many.tsv",
"OrthologuesStats_many-to-one.tsv",
"OrthologuesStats_many-to-many.tsv"
)
# === Function to Read and Process Data ===
read_data_matrix <- function(file_path) {
data <- read.delim(file_path, check.names = FALSE, row.names = 1)
return(as.matrix(data))
}
# === Load Data from Files ===
d11 <- read_data_matrix(paste0(dResults, files[1]))
d1m <- read_data_matrix(paste0(dResults, files[2]))
dm1 <- read_data_matrix(paste0(dResults, files[3]))
dmm <- read_data_matrix(paste0(dResults, files[4]))
# === Schistocerca Species Selection and Order ===
species <- c(
"Sscub_filteredTranscripts", # cubense
"Samer_filteredTranscripts", # americana
"Spice_filteredTranscripts", # piceifrons
"Scanc_filteredTranscripts", # cancellata
"Snite_filteredTranscripts", # nitens
"Sgreg_filteredTranscripts", # gregaria
"Gbima_filteredTranscripts", # Gryllus bimaculatus
"Glong_filteredTranscripts", # Gryllus longicornis
"Asimp_filteredTranscripts", # A. simplex (Stick Insect)
"Brsri_filteredTranscripts", # B. rossius (Stick Insect)
"Daus_filteredTranscripts", # D. australis (Lord Howe Island Stick Insect)
"Pamer_filteredTranscripts", # P. americana (American Cockroach)
"Csecu_filteredTranscripts" # C. secundus (Drywood Termite)
)
# === Map Species to Readable Labels ===
species_labels <- c(
"Sscub_filteredTranscripts" = "cubense",
"Samer_filteredTranscripts" = "americana",
"Spice_filteredTranscripts" = "piceifrons",
"Scanc_filteredTranscripts" = "cancellata",
"Snite_filteredTranscripts" = "nitens",
"Sgreg_filteredTranscripts" = "gregaria",
"Gbima_filteredTranscripts" = "G. bimaculatus",
"Glong_filteredTranscripts" = "G. longicornis",
"Asimp_filteredTranscripts" = "A. simplex",
"Brsri_filteredTranscripts" = "B. rossius",
"Daus_filteredTranscripts" = "D. australis",
"Pamer_filteredTranscripts" = "P. americana",
"Csecu_filteredTranscripts" = "C. secundus"
)
# === Update Row Names ===
rownames(d11) <- species
rownames(d1m) <- species
rownames(dm1) <- species
rownames(dmm) <- species
# === Function to Prepare Data (Self-Comparison Blank) ===
combine_data <- function(d11, d1m, dm1, dmm, species, species_to_plot) {
isp <- which(species == species_to_plot)
dtot <- d11 + d1m + dm1 + dmm
data <- data.frame(
Species = species,
`1:1` = ifelse(species == species_to_plot, 0, d11[isp, ] / dtot[isp, ] * 100),
`1:many` = ifelse(species == species_to_plot, 0, d1m[isp, ] / dtot[isp, ] * 100),
`many:1` = ifelse(species == species_to_plot, 0, dm1[isp, ] / dtot[isp, ] * 100),
`many:many` = ifelse(species == species_to_plot, 0, dmm[isp, ] / dtot[isp, ] * 100)
)
return(data)
}
# === Function to Create and Save Stacked Bar Plot (Vertical) ===
create_plot <- function(data, species_to_plot, output_dir = ortho_dir) {
data_long <- data %>%
pivot_longer(
cols = -Species,
names_to = "Relationship",
values_to = "Proportion"
)
# Rename relationships to match expected levels
data_long$Relationship <- str_replace_all(data_long$Relationship, c(
"X1.1" = "1:1",
"X1.many" = "1:many",
"many.1" = "many:1",
"many.many" = "many:many"
))
# Ensure Relationship column is a factor with consistent levels
data_long$Relationship <- factor(
data_long$Relationship,
levels = c("1:1", "1:many", "many:1", "many:many")
)
# Set Custom Species Order
data_long$Species <- factor(data_long$Species, levels = species, labels = species_labels)
# Generate the Plot
plot <- ggplot(data_long, aes(x = Species, y = Proportion, fill = Relationship)) +
geom_bar(stat = "identity", position = "stack") +
scale_fill_manual(
values = c(
`1:1` = "forestgreen",
`1:many` = "orange",
`many:1` = "purple",
`many:many` = "black"
)
) +
labs(
title = paste("Orthologue multiplicty vs", species_labels[species_to_plot]),
x = "Species",
y = "Proportion (%)",
fill = "Relationship Type"
) +
theme_minimal() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1),
axis.title.x = element_text(size = 12),
axis.title.y = element_text(size = 12)
)+
coord_flip()
# Save the Plot
output_file <- paste0(output_dir, "VerticalStackedBar_", species_labels[species_to_plot], ".pdf")
ggsave(output_file, plot = plot, width = 10, height = 6)
cat("Plot saved as:", output_file, "\n")
}
# === Generate Vertical Plots for Each Schistocerca Species ===
output_dir <- paste0(ortho_dir, "Plots_Polyneoptera/") # Specify output directory
dir.create(output_dir, showWarnings = FALSE)
for (species_to_plot in species) {
data <- combine_data(d11, d1m, dm1, dmm, species, species_to_plot)
create_plot(data, species_to_plot, output_dir)
}Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_cubense.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_americana.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_piceifrons.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_cancellata.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_nitens.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_gregaria.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_G. bimaculatus.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_G. longicornis.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_A. simplex.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_B. rossius.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_D. australis.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_P. americana.pdf Plot saved as: /Users/maevatecher/Documents/GitHub/locust-comparative-genomics/data/orthofinder/Polyneoptera/Results_I2/Plots_Polyneoptera/VerticalStackedBar_C. secundus.pdf library(readr)
# rename for future merging
names(orthogroups)[names(orthogroups) == "Gene"] <- "protein_id"
orthogroups <- orthogroups %>%
mutate(protein_id = str_remove(protein_id, "_p[1-7]$"))
# Optional if you did not rename fasta sequence before
orthogroups$Species <- gsub("_filteredproteome", "", orthogroups$Species)
# Export the table to tab-separated text file (you can change the delimiter if needed)
output_file <- file.path(ortho_dir, "Orthogroups_13species_Jan2025.txt")
# Write the transformed data to the file
write.table(orthogroups,
file = output_file,
sep = "\t",
quote = FALSE,
row.names = FALSE)
proteingene_path <- file.path(ortho_dir, "../../../list/allspecies_protein2geneid.tsv")
proteingeneid <- read_tsv(proteingene_path, col_names = TRUE )
head(proteingeneid)# A tibble: 6 × 4
protein_id GeneID Description Species
<chr> <chr> <chr> <chr>
1 XP_047114676.1 LOC124794980 piggyBac transposable element-derived pro… Schist…
2 XP_047114773.1 LOC124795035 piggyBac transposable element-derived pro… Schist…
3 XP_047117829.1 LOC124798450 uncharacterized protein LOC124798450 Schist…
4 XP_047118606.1 LOC124799109 RNA-binding protein 25-like Schist…
5 XP_047118853.1 LOC124799306 molybdenum cofactor biosynthesis protein … Schist…
6 XP_047118904.1 LOC124799809 molybdenum cofactor biosynthesis protein … Schist…biotype_path <- file.path(ortho_dir, "../../../list/13polyneoptera_geneid_ncbi.csv")
biotypeid <- read_csv(biotype_path, col_names = TRUE )
head(biotypeid)# A tibble: 6 × 10
Accession Begin End Description Symbol GeneID GeneType Transcripts_accession
<chr> <dbl> <dbl> <chr> <chr> <chr> <chr> <chr>
1 NC_06468… 1 66 tRNA-Ile trnI LOC73… tRNA <NA>
2 NC_06468… 70 138 tRNA-Gln trnQ LOC73… tRNA <NA>
3 NC_06468… 138 206 tRNA-Met trnM LOC73… tRNA <NA>
4 NC_06468… 207 1227 NADH dehyd… ND2 LOC73… protein… <NA>
5 NC_06468… 1228 1295 tRNA-Trp trnW LOC73… tRNA <NA>
6 NC_06468… 1288 1350 tRNA-Cys trnC LOC73… tRNA <NA>
# ℹ 2 more variables: protein_id <chr>, Species <chr># Ensure GeneID is treated as a character to avoid mismatches
proteinorthotable <- left_join(orthogroups, proteingeneid, by = "protein_id")
final_orthotable <- left_join(biotypeid, proteinorthotable[, c("Orthogroup","SpeciesID", "GeneID")], by = "GeneID")
output_file <- file.path(ortho_dir, "Orthogroups_genesproteinbiotype_13species_Jan2025.csv")
# Write the table with proper quoting and no row names
write.table(final_orthotable, file = output_file, sep = ",", quote = TRUE, row.names = FALSE, col.names = TRUE)There we have it, the final table with all the corresponding IDs. This table is too big and have all species in it, so we want to reduce to only single copy orthologs and remove entries with other species than Schistocerca for now.
# Filter the final_orthotable to keep only rows with species 'Schistocerca'
filtered_final_orthotable <- final_orthotable %>%
filter(Species %in% c("Schistocerca gregaria", "Schistocerca piceifrons", "Schistocerca americana", "Schistocerca cancellata", "Schistocerca serialis cubense", "Schistocerca nitens"))
# Optionally, save the filtered data
output_file <- file.path(ortho_dir, "Orthogroups_genesprotein_Schisto_Jan2025.txt")
write.table(filtered_final_orthotable, file = output_file, sep = "\t", quote = FALSE, row.names = FALSE)
# Step 1: Read the single copy orthologs table
single_copy_orthologs_path <- file.path(ortho_dir, "Orthogroups/Orthogroups_SingleCopyOrthologues.txt")
single_copy_orthologs <- read.table(single_copy_orthologs_path, header = FALSE, stringsAsFactors = FALSE)
# Step 2: Ensure the column name for orthogroups matches in both data frames
# If necessary, rename the column in single_copy_orthologs to match
colnames(single_copy_orthologs) <- c("Orthogroup") # Replace with the actual name if different
# Step 3: Perform the intersection
scopy_final_orthotable <- final_orthotable[final_orthotable$Orthogroup %in% single_copy_orthologs$Orthogroup, ]
# Step 4: Optionally, save the filtered table
output_file <- file.path(ortho_dir, "SingleCopyOrthogroups_genesprotein_13species_Jan2025.txt")
write.table(scopy_final_orthotable, file = output_file, sep = "\t", quote = FALSE, row.names = FALSE)References
If you use this script, and orthologr please cite:
Drost et al. 2015. Evidence for Active Maintenance of Phylotranscriptomic Hourglass Patterns in Animal and Plant Embryogenesis. Mol. Biol. Evol. 32 (5): 1221-1231. doi:10.1093/molbev/msv012
If you use Transdecoder, please cite it as:
Haas, B., and A. Papanicolaou. “TransDecoder.” (2017).
If you use Orthofinder, please cite it:
OrthoFinder’s orthogroup and ortholog inference are described here:
Emms, D.M., Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol 16, 157 (2015).
Emms, D.M., Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20, 238 (2019).
If you use the OrthoFinder species tree then also cite:
Emms D.M. & Kelly S. STRIDE: Species Tree Root Inference from Gene Duplication Events (2017), Mol Biol Evol 34(12): 3267-3278.
Emms D.M. & Kelly S. STAG: Species Tree Inference from All Genes (2018), bioRxiv https://doi.org/10.1101/267914.
Please also cite MAFFT:
K. Katoh, K. Misawa, K. Kuma, and T. Miyata. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14): 3059-3066.
sessionInfo()R version 4.4.1 (2024-06-14)
Platform: aarch64-apple-darwin20
Running under: macOS 15.3
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Asia/Tokyo
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] readr_2.1.5 tibble_3.2.1 tidyr_1.3.1 stringr_1.5.1
[5] ggplot2_3.5.1 dplyr_1.1.4 treeio_1.28.0 ggtree_3.12.0
[9] cogeqc_1.8.0 kableExtra_1.4.0 knitr_1.49
loaded via a namespace (and not attached):
[1] tidyselect_1.2.1 viridisLite_0.4.2 vipor_0.4.7
[4] farver_2.1.2 Biostrings_2.72.1 fastmap_1.2.0
[7] lazyeval_0.2.2 promises_1.3.2 digest_0.6.37
[10] lifecycle_1.0.4 tidytree_0.4.6 magrittr_2.0.3
[13] compiler_4.4.1 rlang_1.1.4 sass_0.4.9
[16] tools_4.4.1 utf8_1.2.4 igraph_2.1.2
[19] yaml_2.3.10 labeling_0.4.3 bit_4.5.0.1
[22] plyr_1.8.9 xml2_1.3.6 aplot_0.2.4
[25] workflowr_1.7.1 withr_3.0.2 purrr_1.0.2
[28] BiocGenerics_0.50.0 grid_4.4.1 stats4_4.4.1
[31] git2r_0.35.0 colorspace_2.1-1 scales_1.3.0
[34] cli_3.6.3 rmarkdown_2.29 crayon_1.5.3
[37] ragg_1.3.3 generics_0.1.3 rstudioapi_0.17.1
[40] tzdb_0.4.0 httr_1.4.7 reshape2_1.4.4
[43] ggbeeswarm_0.7.2 ape_5.8-1 cachem_1.1.0
[46] zlibbioc_1.50.0 parallel_4.4.1 ggplotify_0.1.2
[49] XVector_0.44.0 vctrs_0.6.5 yulab.utils_0.1.9
[52] jsonlite_1.8.9 hms_1.1.3 gridGraphics_0.5-1
[55] IRanges_2.38.1 patchwork_1.3.0 S4Vectors_0.42.1
[58] bit64_4.5.2 beeswarm_0.4.0 systemfonts_1.1.0
[61] jquerylib_0.1.4 glue_1.8.0 stringi_1.8.4
[64] gtable_0.3.6 later_1.4.1 GenomeInfoDb_1.40.1
[67] UCSC.utils_1.0.0 munsell_0.5.1 pillar_1.10.1
[70] htmltools_0.5.8.1 GenomeInfoDbData_1.2.12 R6_2.5.1
[73] textshaping_0.4.1 rprojroot_2.0.4 vroom_1.6.5
[76] evaluate_1.0.3 lattice_0.22-6 httpuv_1.6.15
[79] ggfun_0.1.8 bslib_0.8.0 Rcpp_1.0.14
[82] svglite_2.1.3 nlme_3.1-166 whisker_0.4.1
[85] xfun_0.50 fs_1.6.5 pkgconfig_2.0.3